
Lead
Changes in selectivity brought about by modifying the composition of the mobile phase have been discussed up until this point. This method is typically the most practical and economical way to alter selectivity. Alternately, relying on changes in column selectivity to control the separation is a strategy that is typically reserved until all other mobile phase alternatives have been exhausted. As with alterations to the selectivity of the mobile phase, chromatographers should make substantial alterations to the selectivity of the column to derive the most benefit from this parameter. Consequently, switching from one brand of C18 column to another or from a Cl8 column to a C8 column will not be particularly fruitful. Variations in selectivity may result from these kinds of modifications, but they are typically minor in comparison to modifications to the stationary-phase chemistry.
Column Selectivity
Traditionally, three column types – Cl8 or C8, Cyano, and phenyl – are chosen for reversed-phase separations. Figures 1 and 2 depict the variations in selectivity between a C8, Cyano, and phenyl bonded phase for two distinct samples. Figure 1 represents a group of substituted benzoic acids.
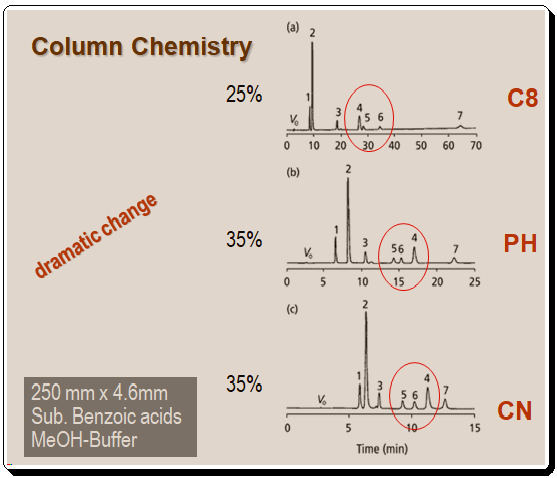
The most notable difference is in the retention order of the 4-5-6 triplet, with compound 4 appearing first in the C8 column and last in the other two. Additionally, the peak spacing between peaks 1 and 2 varies significantly. A mixture of herbicides was used for the separations in Figure 2. Peaks 1 and 2 converge on the cyano column, and the separation between peaks 6 and 7 is also diminished. On these columns, no retention order changes are observed for the herbicide mixture.
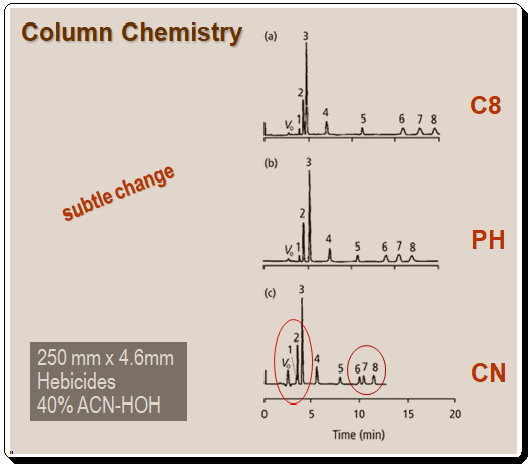
It is evident that changes in column chemistry can result in dramatic changes in selectivity, as in the case of the benzoic acids in Figure 1, or much more subtle changes, as in the case of the herbicide mixture in Figure 2. It is impossible to accurately predict changes in selectivity between columns, and there is no software available to assist in fine-tuning selectivity changes, such as the software used for mobile-phase modifications; therefore, column changes tend to be based on trial-and-error. Before exploring column selectivity, it is suggested to attempt the simpler and often more effective mobile-phase changes.
Other variables
The focus of the discussion thus far has been on the variables that have the greatest impact on the selectivity of a separation. Many additional variables can be investigated. For instance, ion-pair reagents are frequently used to improve selectivity, particularly when a mixture of acids and bases is present in the sample, but ion pairing is typically more problematic than pH control. Chromatographers can gain selectivity by utilizing unique chemical characteristics of the sample, such as the presence of chelators, chiral compounds, or shape differences. When a good effort has been made to obtain a separation using reversed-phase variables, workers may wish to investigate chromatography techniques such as ion exchange, normal phase, and size exclusion. The reference number 7 is a useful resource for developing methods for these and other samples.
Conclusions
By the time chromatographers have experimented with mobile-phase solvents, pH, temperature, column type, and possibly ion pairing, they will have most likely achieved a satisfactory reversed-phase separation. Occasionally, the selectivity or peak spacing is adequate, but the peaks are not quite baseline separated. In other instances, the peaks may be overly separated, which indicates time wastage. At this point in the method development procedure, it is appropriate to determine whether modifying the column size, mobile-phase flow rate, or packing particle diameter will improve the results. These modifications to column parameters will be the focus of the next blog.
For detailed treatment of any of the above concepts consult books/article suggested in the “Further reading” section.
Acknowledgment
The author wishes to acknowledge the use of simulated chromatogram from reference 3, has no intellectual property rights over the material, and has resorted to using it for educational and training purposes.
Disclaimer
Due to the field’s quick development, there may have been some unintentional omissions or inadequate technical specifications. If any omissions or updates needing to be made are added, we would be pleased to include them. To draw attention to it, kindly write a comment. All technical specifications found here are reproduced as-is from the manufacturer’s literature and might have changed at the time of publication. All information given here is for furthering science and cannot be construed as professional advice.
Further reading
1. Lloyd R. Snyder, Joseph J. Kirkland, Joseph L. Glajch, Practical HPLC Method Development, pages. 600-615, 1997, John Wiley & Sons, Inc. ISBN 0-471-00703-X
2. Lloyd R. Snyder, Joseph J. Kirkland, John W. Dolan Introduction to Modern Liquid, Chromatography, 3rd ed., Wiley 2009. ISBN: 978-0-470-16754-0
3. John W. Dolan, LCGC, 2000, 18(3), 286-293.
4. John W. Dolan, LCGC, 2000, 18(2), 118-125.
5. John W. Dolan, LCGC, 2000, 18(1), 28-32.
6. John W. Dolan, LCGC, 1999, 17(12), 1094-1097.
7. John W. Dolan and Lloyd R. Snyder, Troubleshooting LC systems: A comprehensive approach to troubleshooting LC equipment and separations, 1989, Humana Press, Inc., New Jersey. ISBN 0-89603-151-9.
8. Henrik Rasmussen (Editor), Satinder Ahuja (Editor), HPLC Method Development for Pharmaceuticals: Volume 8 (Separation Science and Technology), 2007, Academic Press Inc, ISBN-13 : 978-3527331291.
9. https://chiralpedia.com/blog/selecting-the-tools
10. https://chiralpedia.com/blog/measuring-quality-of-chromtogram-tools-1.0/
11.. https://chiralpedia.com/blog/measuring-quality-of-chromatogram-tools-2-0/
12. https://chiralpedia.com/blog/measuring-quality-of-chromatogram-tools-3-0/
13. https://chiralpedia.com/blog/controlling-selectivity-solvent-role-1-0/
14. https://chiralpedia.com/blog/controlling-selectivity-solvent-role-2-0/
15. https://chiralpedia.com/blog/controlling-selectivity-additional-factors-1-0-ph/
16. https://chiralpedia.com/blog/controlling-selectivity-additional-factors-2-0-temperature/
