
Lead
This is in continuation to my earlier post <https://chiralpedia.com/blog/controlling-selectivity-solvent-role-1-0/> where we discussed the role solvent, first approach – varying solvent strength, in controlling retention factor and selectivity of chromatographic separation. In this post the focus is on the second approach, blending of solvents, to arrive at a good separation of difficult-to-separate peak pairs in HPLC method development.
Second Approach: Blend solvents
Òne can find suitable separation conditions for many samples simply by adjusting the solvent strength. However, for some other analytes no acetonitrile concentration will result in a good separation. At this point, you ought to change your strategy for changing α. Switching to another organic solvent in the mobile phase is a potent way to change α for many samples. An approach to illustrate this approach is depicted in the simulate chromatogram for the sample of Figure 4.
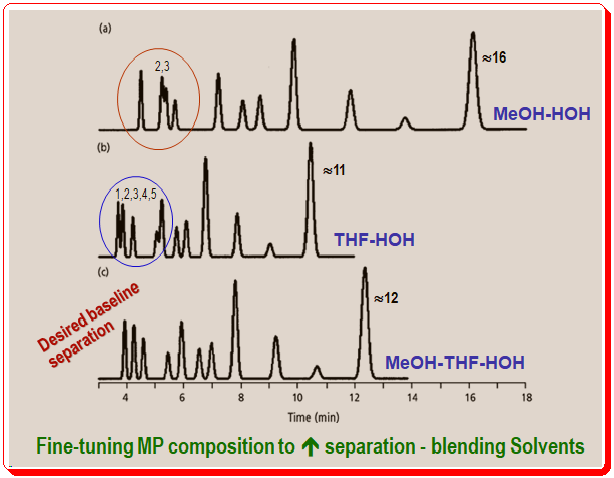
Acetonitrile and water mobile phases were unable to separate the sample of Figure 4 as desired. The next ideal choice from the mobile phase solvents is methanol.
The best separation is shown in Figure 4a using methanol-water (55% methanol). Peaks 2 and 3 (marked by red oval circle) are poorly separated, but the majority of the peaks in the chromatogram have satisfactory resolution. Another solvent was tried to adjust the concentration of solvent B without success. Tetrahydrofuran (THF) was used in place of methanol for the separation in Figure 4b, and this figure demonstrates that the resolution of the run’s later peaks is satisfactory. The initial portion of the run, though, is still problematic. The resolution is poor for peaks 1 and 2, and 4 and 5 (marked by blue oval circle). Again, it is found that adjusting the concentration of solvent B didn’t improve the situation. Acetonitrile, methanol, and tetrahydrofuran all failed to produce a successful separation of this sample. However, the separations of Figure 4 show another cautious that successful chromatographers ought to practice. The critical peak pairs, or those that are the most difficult to resolve, vary between runs in Figures 4a and 4b. Possibly an intermediate set of conditions should produce peak separations and retention times that fall between those seen for the best methanol-water and tetrahydrofuran-water conditions, just as was the case for the sample of Figure 3 (consult earlier post -<https://chiralpedia.com/blog/controlling-selectivity-solvent-role-1-0/>). On similar lines, the separation of sample in Figure c, was achieved by blending the isocratic solvents of Figure 4a and 4b. All chromatogram peaks have the desired baseline resolution under these circumstances.
Sum-up
In reversed-phase LC separations, selectivity is greatly influenced by the organic solvent. Few recommendations are listed below that you should keep in mind based on the aforementioned examples. First, alter the solvent B concentration (acetonitrile is a good starting point) until all of the peaks fall within the 1 < k < 20 region. Watch the movement of individual peaks to determine when fine-tuning the concentration of solvent B is appropriate. In general, resolution will rise with increasing retention. Try methanol and run the optimization experiments again if acetonitrile doesn’t work; if methanol doesn’t work, try tetrahydrofuran. As you make these adjustments, be mindful of relative peak movements because shifts in selectivity between conditions frequently point to the need for an intermediary condition or solvent blend to separate difficult-to-separate peak pairs.
Modifying % B – Guidelines:

Although solvent type and strength have a significant impact on chromatographic selectivity, they are not the only factors that can be considered. Other factors/variables that can be used to control selectivity will be highlighted in the forthcoming post.
For a detailed treatment of any of the above concepts consult books/articles suggested in the “Further reading” section.
Acknowledgment
The author wishes to acknowledge the use of simulated chromatograms from reference 3, has no intellectual property rights over the material, and has resorted to using it for educational and training purposes.
Disclaimer
Due to the field’s quick development, there may have been some unintentional omissions or inadequate technical specifications. If any omissions or updates needing to be made are added, we would be pleased to include them. To draw attention to it, kindly write a comment. All technical specifications found here are reproduced as-is from the manufacturer’s literature and might have changed at the time of publication. All information given here is for furthering science and cannot be construed as professional advice.
Further reading
1. Lloyd R. Snyder, Joseph J. Kirkland, Joseph L. Glajch, Practical HPLC Method Development, pages. 600-615, 1997, John Wiley & Sons, Inc. ISBN 0-471-00703-X
2. Lloyd R. Snyder, Joseph J. Kirkland, John W. Dolan Introduction to Modern Liquid, Chromatography, 3rd ed., Wiley 2009. ISBN: 978-0-470-16754-0
3. John W. Dolan, LCGC, 2000, 18(2), 118-125.
4. John W. Dolan, LCGC, 2000, 18(1), 28-32.
5. John W. Dolan, LCGC, 1999, 17(12), 1094-1097.
6. John W. Dolan and Lloyd R. Snyder, Troubleshooting LC systems: A comprehensive approach to troubleshooting LC equipment and separations, 1989, Humana Press, Inc., New Jersey. ISBN 0-89603-151-9.
7. Henrik Rasmussen (Editor), Satinder Ahuja (Editor), HPLC Method Development for Pharmaceuticals: Volume 8 (Separation Science and Technology), 2007, Academic Press Inc, ISBN-13 : 978-3527331291.
8. https://chiralpedia.com/blog/selecting-the-tools
9. https://chiralpedia.com/blog/measuring-quality-of-chromtogram-tools-1.0/
10.. https://chiralpedia.com/blog/measuring-quality-of-chromatogram-tools-2-0/
11. https://chiralpedia.com/blog/measuring-quality-of-chromatogram-tools-3-0/
12. https://chiralpedia.com/blog/controlling-selectivity-solvent-role-1-0/
