“Measuring handedness – tools and techniques that bring chirality into focus”
Introduction
Ensuring the correct stereochemistry and measuring stereochemical purity is a crucial aspect of pharmaceutical quality control and research. This part covers the major analytical techniques used to distinguish and quantify enantiomers and diastereomers in drug substances and products.
Key techniques include:
- Chiral Chromatography: High-Performance Liquid Chromatography (HPLC) and Gas Chromatography (GC) on chiral stationary phases – the workhorses for separating enantiomers.
- Nuclear Magnetic Resonance (NMR) Spectroscopy: Methods like using chiral solvating agents or derivatizing agents to split enantiomer signals, as well as NOE and coupling constant analysis for stereochemical assignments.
- X-ray Crystallography: The definitive method for determining absolute configuration when a suitable crystal can be obtained (especially if heavy atoms present for anomalous dispersion).
- Optical Rotation and Circular Dichroism: Classical polarimetric measurements for optical activity and Circular Dichroism (CD) spectroscopy, including optical rotatory dispersion (ORD) and CD in UV (electronic CD) and IR (Vibrational CD), to get stereochemical information.
- Mass Spectrometry Chirality: Emerging techniques like chiral MS (e.g., using kinetic method or forming diastereomeric complexes and measuring differences) – more specialized.

We’ll discuss how these techniques are applied in practice: e.g., during process development, a chiral HPLC method is developed to monitor enantiomeric excess of an API; during formulation QC, the final product is tested to ensure no racemization occurred on shelf. For NMR, we illustrate how a chiral shift reagent (like Eu(hfc)_3) can cause separate NMR signals for enantiomers or how Mosher’s method is used to determine absolute configuration of stereocenters in a molecule by NMR. A comparative table of techniques with their scope, sensitivity, and limitations will be sketched as a visual aid.
Chiral Chromatography (HPLC and SFC)
HPLC: Chiral HPLC is often considered the gold standard for enantiomer separation in analytical labs. Columns containing a chiral stationary phase (CSP) are used. Common CSP types: – Polysaccharide derivatives: e.g., cellulose or amylose tris(phenylcarbamate) derivatives (Chiralpak AD, Chiralcel OD, etc.) are very versatile. They work by multiple weak interactions – enantiomers fit differently into the chiral grooves of these polymeric phases. – Cyclodextrin-based: especially in GC, but also HPLC; cyclodextrins can resolve certain chiral analytes by inclusion complex formation. – Protein-based: e.g., a column with immobilized bovine serum albumin can separate some enantiomers (the protein’s chiral pockets bind one enantiomer more strongly). – Macrocyclic glycopeptides: like vancomycin-based phases (Chirobiotic V) that can separate many drug enantiomers (the multiple chiral centers and binding sites in vancomycin provide chiral discrimination). – Ligand exchange columns: e.g., a copper(II) complex of L-proline on a column, which can separate amino acid enantiomers by forming transient diastereomeric complexes.
Chiral HPLC is so powerful that for many drugs the enantiomeric purity is just tested by HPLC with UV detection. For instance, the USP monograph might say: inject sample into a chiral column and the undesired enantiomer must be <0.1%. Limits are usually stringent (since the distomer could be harmful or just considered an impurity). For a product like levofloxacin (the S-enantiomer of ofloxacin), the EP/USP require verifying that the R-enantiomer is under a small percentage.
Chiral SFC (Supercritical Fluid Chromatography) is similar to HPLC but uses supercritical CO2 as the main mobile phase with co-solvents. It’s often faster and with high resolution for chiral separations and is used preparatively too (with automated fraction collection). It’s become popular in pharma for rapid chiral method development and even purification. Many chiral stationary phases are cross-compatible with SFC. SFC’s advantage: lower viscocity -> higher flow, plus easy solvent removal (CO2 evaporates).
Chiral GC: Many small chiral molecules (volatiles like flavors, pheromones) are analyzed by GC using chiral phases (often cyclodextrin-based capillaries). For drugs, GC might be used if the compound or its derivative is volatile enough. For example, an amino acid might be derivatized to an ester and then separated on a Chirasil-Val (a valine-derived stationary phase) GC column. But most pharma compounds are more amenable to HPLC.
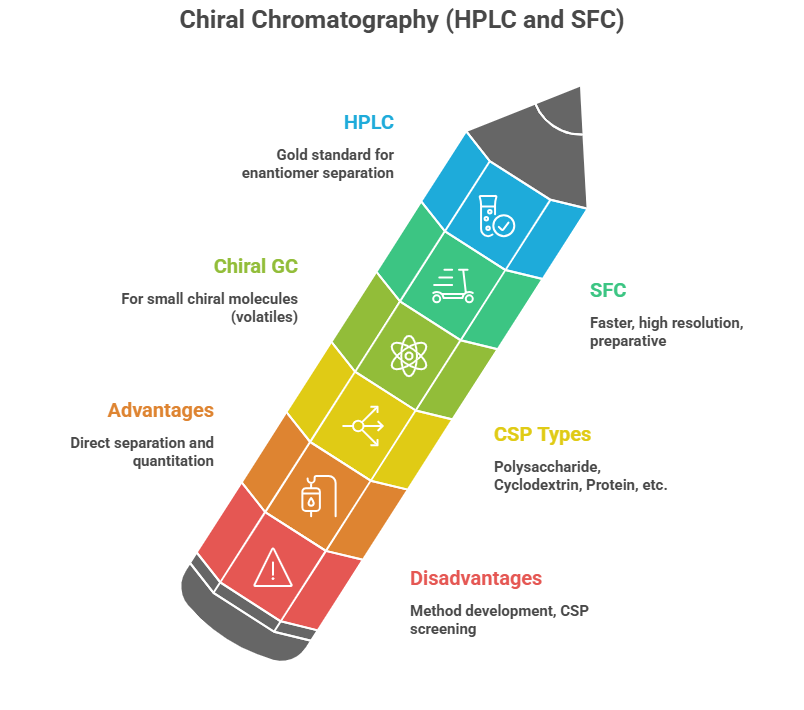
Advantages/Disadvantages: – Chromatography provides direct separation and quantitation. HPLC is widely applicable (works for a broad range of compounds). – It requires method development and a good chiral column match – sometimes screening multiple CSPs is needed to find one that separates a particular enantiomer pair (some might co-elute on one column and separate on another). – For QC, robust chiral HPLC methods are established, often validated as per ICH. – Sensitivity: If the compound has UV absorbance, detection is straightforward. For compounds without chromophores, detectors like polarimetric detectors or mass spec can be used, but typically, most drugs have UV or can be derivatized to have one.
For a detailed perspective, you may like this series of blog articles: <#chiral_chromatography>
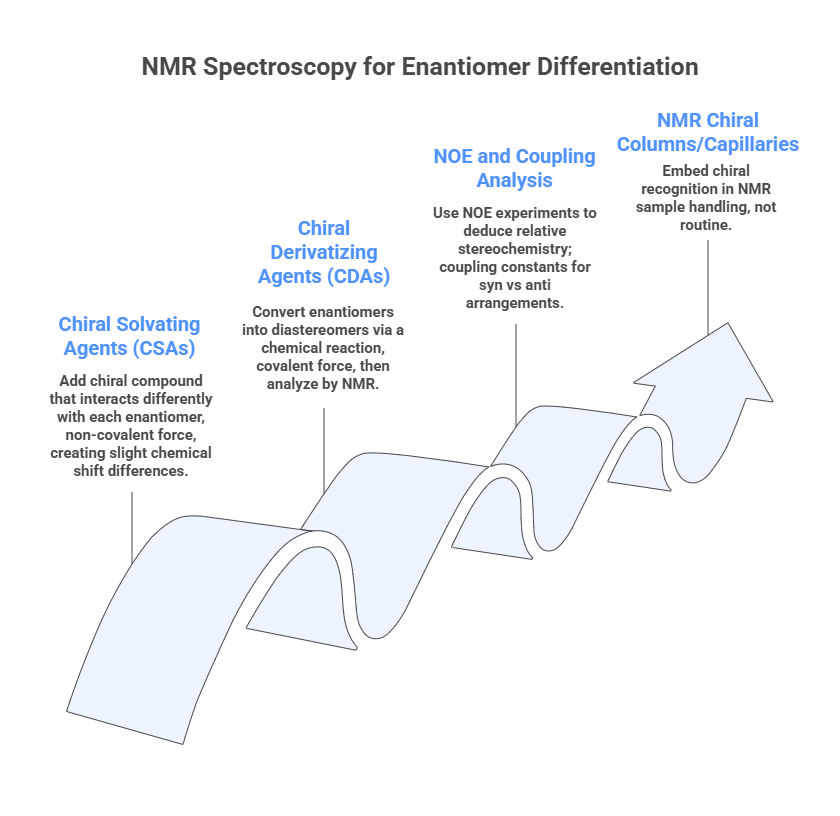
NMR Spectroscopy
By itself, NMR of a racemic mixture in an achiral solvent will show identical spectra for enantiomers (their signals overlap since they are chemically and magnetically equivalent in symmetric environment). To distinguish them, one must break that symmetry: – Chiral Solvating Agents (CSAs): Add a chiral compound that interacts (through transient or non-covalent interaction) differently with each enantiomer, creating slight chemical shift differences diastereomeric complexes form). E.g., Eu(hfc)_3 (Europium tris-[3-(heptafluoropropylhydroxymethylene)-(+)-camphorate]) is a chiral shift reagent that often resolves NMR signals of enantiomers. Another CSA is Pirkle’s alcohol or Mosher’s salt. A simple one: 2,2,2-trifluoro-1-(9-anthryl)ethanol is a CSA for certain compounds. This method is used to measure enantiomeric excess by integrating separated signals of enantiomeric protons. It’s useful if one has a mixture and a quick NMR check is needed. For example, to test if an asymmetric synthesis gave 90% ee, one could add a dash of CSA and see separate NMR peaks for each enantiomer.
- Chiral Derivatizing Agents (CDAs): Convert the enantiomers into diastereomers via a chemical reaction (covalent bond), then analyze by NMR (or GC). A famous one is Mosher’s acid chloride (α-methoxy-α-trifluoromethylphenylacetic acid chloride). Suppose you have a chiral secondary alcohol as a racemate. React both enantiomers with Mosher’s chloride to form esters. These diastereomeric esters will show distinct NMR signals (especially the methoxy and trifluoromethyl groups can serve as probes). The differences in chemical shifts can be used to determine which configuration the alcohol was (the Mosher method provides a rule based on the observed Δδ values). Also, integration of certain signals gives ratio of diastereomers => enantiomeric ratio originally. Mosher’s method is widely taught for assigning absolute config of alcohols and amines.
- NOE and coupling analysis: For diastereomers or conformers, one can use NOE (Nuclear Overhauser Effect) experiments to deduce relative stereochemistry in a molecule (like in a ring, cis vs trans substituents will have distinct NOE patterns). For acyclic systems, coupling constants (vicinal J) can follow Karplus relationships differently for syn vs anti arrangements. E.g., differentiate erythro vs threo diastereomers by NMR coupling. This is more for structure elucidation rather than quantifying mixtures.
- NMR chiral columns/capillaries: There’s research on embedding chiral recognition in NMR sample handling (like chiral liquid crystals that cause chemical shift differences in enantiomers), but it’s not routine.
NMR’s advantage is that it’s a complementary technique: one can both identify and quantify in one go if the differences are clear. However, it’s generally less sensitive than HPLC for minor impurity detection (HPLC can detect 0.1% enantiomeric impurity with good method, whereas NMR might struggle to see 0.1% of one enantiomer due to sensitivity). So NMR is more for moderate ratios or structural assignment.
X-ray Crystallography
If a compound can be crystallized, X-ray diffraction can determine its 3D structure. However, to get the absolute configuration, you typically need either: – A heavy atom in the molecule (or co-crystallize with a heavy-atom containing reference) to utilize anomalous dispersion (the Bijvoet differences allow determining absolute structure). – Or comparing to a known reference in the crystal (like crystallize with a known chiral counterion). For example, crystallizing a new chiral drug’s salt with an L-tartaric acid can tell you how it oriented relative to the known L configuration.
X-ray is the ultimate proof; many drugs have an X-ray structure in the NDA package that confirms the molecule’s stereochemistry. It’s also used to confirm stereochemistry of complex poly-chiral molecules (like a macrocyclic drug). One limitation: not all molecules crystallize easily, and sometimes the absolute config determination on light-atom molecules (like only C, H, O, N) is challenging if there’s no significant anomalous scatterers. Labs may deliberately add a heavy-atom label to solve that if needed.
One interesting use: historically, (+)- and (–)-tartaric acid’s absolute configs were unknown until Bijvoet in 1949 used X-ray (with anomalous scattering from heavy atoms) on a tartrate salt to determine which hand was which. That was the first absolute config determination by X-ray – a breakthrough.
In QC, X-ray isn’t a routine test for every batch (too slow/cumbersome), but it’s there for initial proof-of-structure.
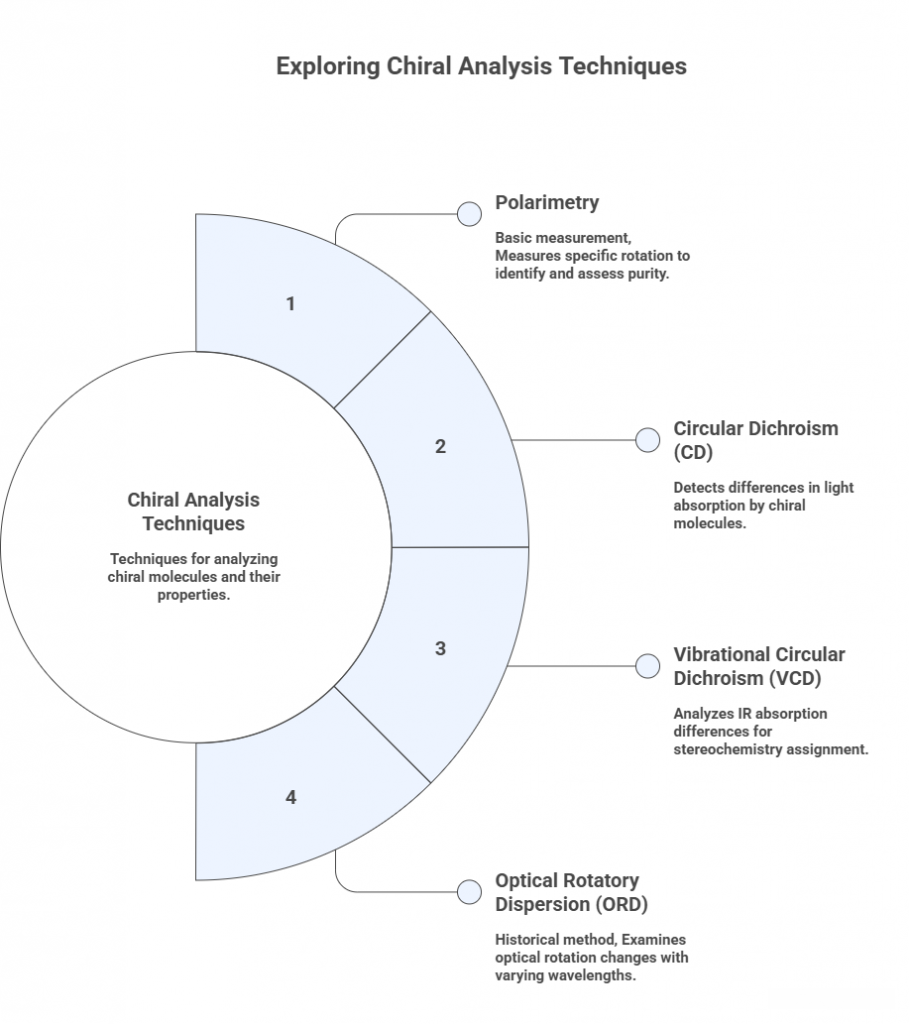
Circular Dichroism (CD) and Optical Rotation
– Polarimetry: We’ve discussed [α] in Part 2. In QC, a drug’s specific rotation can serve as an identity test or quality parameter. E.g., the USP might say [α]_D should be between +25° to +27° for a given enantiopure substance, indicating it’s the right enantiomer and sufficiently pure (a deviant [α] could indicate presence of opposite enantiomer or other impurity). However, polarimetry is a bulk measure and might not catch low-level impurities (one could not easily detect 5% of opposite enantiomer by polarimeter if the [α] is not extremely precise). Polarimetry is simple and nondestructive, so it’s still used as a quick check.
- Circular Dichroism (Electronic): CD measures the difference in absorption of left vs right circularly polarized light by a chiral molecule. It gives a spectrum that can be characteristic of the stereochemistry (the shape of the CD curve is related to stereochemical arrangement of chromophores). CD is particularly useful for molecules with chromophores in chiral environments (like many drugs have UV absorbance). It can be used to assign absolute config by comparing the experimental CD spectrum to literature or calculated spectra. For example, for a complex natural product, chemists sometimes do CD and compare to known analogs to assign config. For small drug molecules, CD is less common in routine QC, but is used in research to confirm assignments.
One specialized form: Vibrational Circular Dichroism (VCD), which looks at IR absorption difference for circular polarizations. It can be used to determine absolute config by comparison with quantum chemical predictions of VCD spectra. VCD has become more popular in academic labs for stereochemistry assignment of flexible molecules where X-ray isn’t available.
- ORD (Optical Rotatory Dispersion): Variation of optical rotation with wavelength. ORD curves can have characteristic Cotton effects. CD is essentially the derivative of ORD (both are related phenomena). Historically used, but nowadays CD is directly measured.
Applications in Pharma QC and Submission
When filing an NDA, companies include methods to test enantiomeric purity. The FDA expects a validated chiral HPLC/GC method if any possibility of undesired enantiomer being present. ICH threshold: often if an enantiomeric impurity is above 0.1-0.2%, it should be controlled or justified. For some drugs, an ICH Q3C impurity analysis includes “other enantiomer” as a specified impurity. E.g., for levofloxacin, the limit for dextrofloxacin (the other enantiomer) might be something like <0.5%. If an enantiomer has known toxicity, the limit might be tighter.
Case examples:
– The NSAID drug ibuprofen (in some markets dexibuprofen is sold) – quality control of dexibuprofen ensures R-ibuprofen is below some threshold. – Thalidomide: if it were single-enantiomer (not done because of in vivo racemization), one would need to ensure not significant racemization occurred in formulation or storage. – Ephedrine/Pseudoephedrine: these are diastereomers and also enantiomeric pairs. In production of L-pseudoephedrine, one ensures no D-pseudoephedrine contamination (which is less active). – Beta-blockers like timolol are often marketed as single enantiomers now; the other enantiomer is just an impurity to be limited.
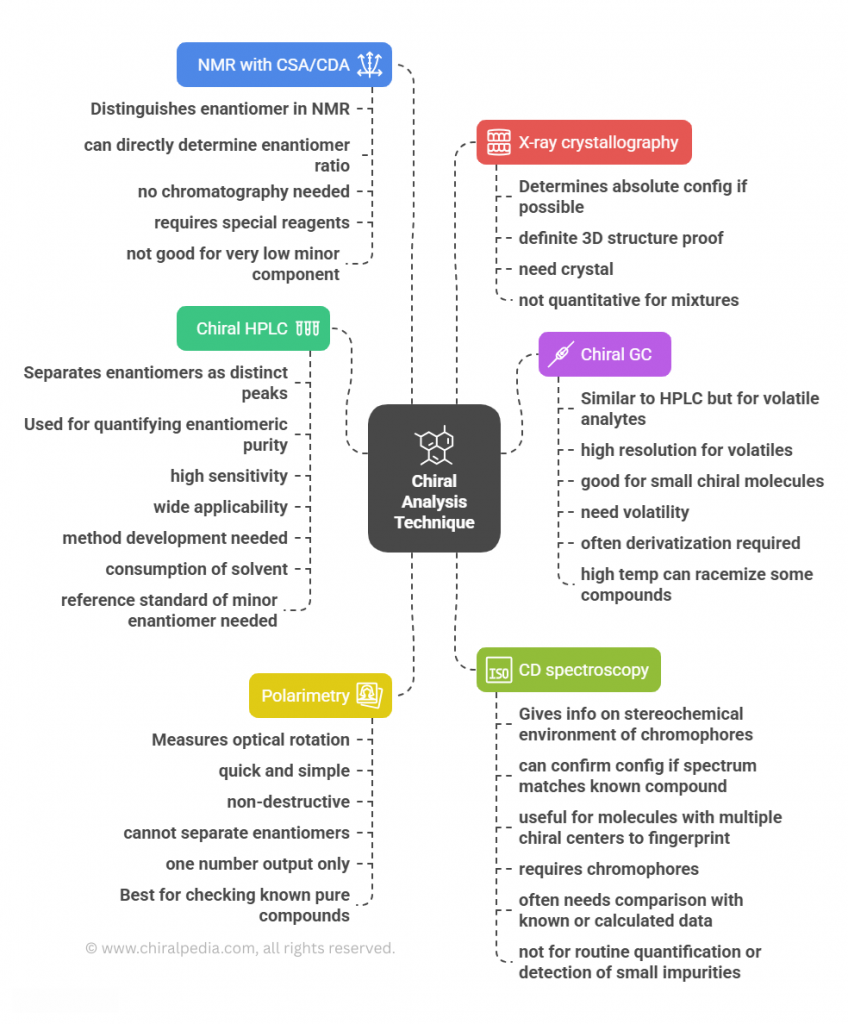
Chiral Analysis Made Visual!
Understanding the landscape of chiral analytical tools can feel overwhelming—different techniques, different capabilities, and unique pros and cons. To make it easier, we’ve created a visual mind map that compares the most widely used methods side by side.
It highlights what each tool measures, its typical applications, strengths, and limitations, giving you a clear, big-picture view. Whether you’re a student, researcher, or industry professional, this snapshot will help you quickly grasp how these techniques complement each other in practice.
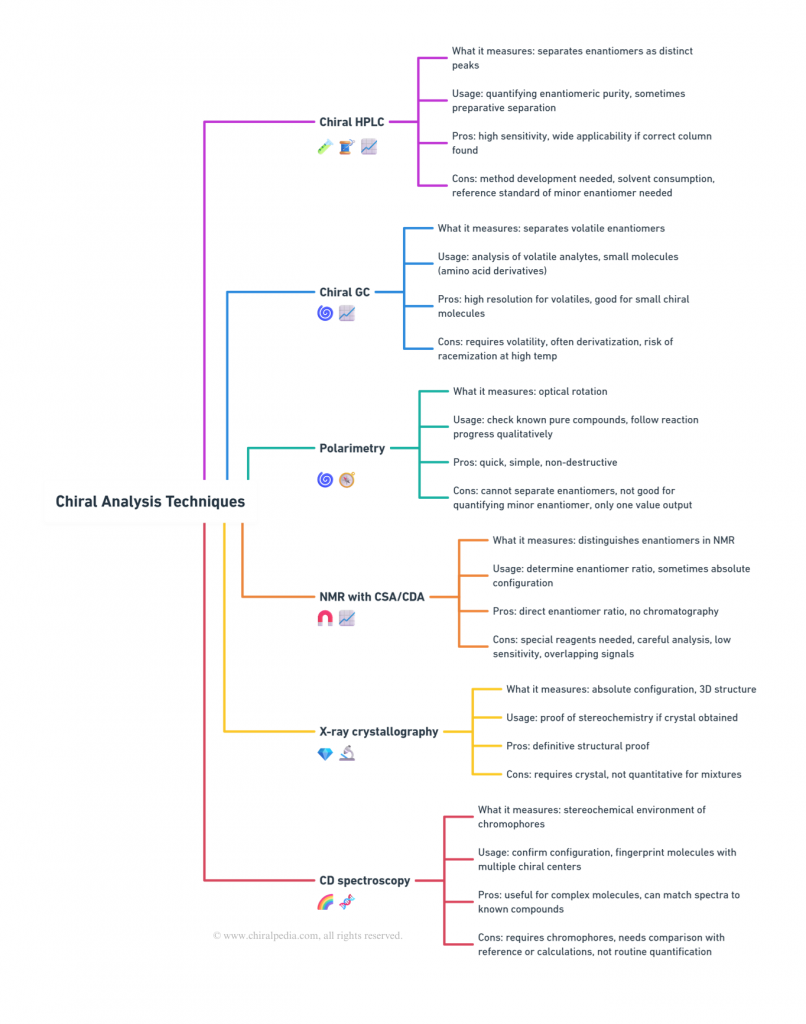
Summary (Part 7)
– A variety of analytical techniques exist to tackle the challenges of chirality in analysis. Chiral HPLC is the most commonly employed for quantitative determination of enantiomeric purity, valued for its precision and reliability in both development and QC labs. – NMR methods complement chromatography, providing insights into stereochemistry and sometimes allowing determination of configuration or purity without chromatographic separation by using chiral agents or derivatization. NMR is also how chemists often initially assign relative stereochemistry in molecules, which is then locked down by X-ray or CD. – X-ray crystallography remains the ultimate arbiter of absolute configuration for new chiral compounds – a single crystal can reveal the stereochemistry definitively (assuming anomalous dispersion data). It’s a key part of structural elucidation for drug substances (especially novel structures). – Optical rotation is an old but still relevant technique – a quick [α]_D measurement can confirm that, say, a batch of L-amino acid is indeed levorotatory and within spec. It’s less detailed than modern methods but provides continuity with historical data (pharmacopoeias often list [α] values). – Analytical methods are often orthogonal – e.g., you might do polarimetry and chiral HPLC on the same sample; if both match expectations, confidence is high. – Regulatory demands ensure that these methods are validated: e.g., chiral HPLC must be shown to accurately quantify low levels of the opposite enantiomer (linearity, LOD/LOQ, etc.). – Analytical techniques also play a role in monitoring stereochemical stability: e.g., stress testing a drug in solution to see if it racemizes under heat/pH (analyzed by chiral chromatography to see if new peak appears). If a drug racemizes easily, that might require special handling or label statements. – Newer technology: chiral detectors (like circular dichroism detectors inline with HPLC to identify eluted enantiomers by their CD spectrum), or mass spec methods for chiral analysis (derivatize with a chiral reagent that adds a mass tag and forms diastereomers that separate by MS) – these are evolving fields.
Suggested Reading
“Chiral Separations by HPLC” by Allenmark or “A Practical Guide to Chiral Separation” (texts on method development for chiral HPLC, including case studies from pharma analysis).
L. Pasteur’s original findings (for historical reference on optical rotation and manual resolution) – not directly practical now but interesting perspective.
https://en.wikipedia.org/wiki/Chiral_analysishttps://www.canada.ca/en/health-canada/services/drugs-health-products/drug-products/applications-submissions/guidance-documents/chemical-entity-products-quality/guidance-industry-stereochemical-issues-chiral-drug-development.html
ACS Chiral Separations Review (2019) – e.g., Maier, N. M., et al. (2001), Journal of Chromatography A, 906, 3-33 (a bit older but thorough on CSPs).
M. R. Luderer and R. L. Powell, “Use of NMR Chiral Solvating Agents in Pharmaceutical Analysis,” American Pharmaceutical Review, discusses CSA method usage in pharma labs.
USP General Chapter <781> Optical Rotation – outlines how to do polarimetry, since pharma labs still reference this for certain chiral excipients or APIs (like in amino acid analysis).
https://en.wikipedia.org/wiki/Chiral_analysis
