“Crafting molecules with precision – the art of stereochemical synthesis”
Introduction
Having seen why the correct stereochemistry is crucial for drug efficacy and safety, the next challenge is how to obtain the desired stereoisomer. This part covers strategies and methods in stereoselective and stereospecific synthesis. We clarify the terminology: a stereoselective reaction preferentially yields one stereoisomer over others (e.g., one enantiomer or one diastereomer is favored), whereas a stereospecific reaction produces different stereoisomeric products from different stereoisomeric reactants (the mechanism links the stereochemistry of starting materials to that of products in a fixed way). In practice, most “asymmetric synthesis” in drug chemistry is stereoselective – aiming to maximize yield of the desired enantiomer or diastereomer.
This section will explore common strategies:
- Chiral pool and chiral auxiliaries – using existing chirality from nature or temporary stereocontrolling groups.
- Chiral reagents – reagents that impart asymmetry during a reaction.
- Chiral catalysts – both metal-based (e.g., organometallic complexes with chiral ligands) and organocatalysts (small organic chiral catalysts).
- Stereoselective enzymatic syntheses – nature’s catalysts for asymmetric transformations.
We will illustrate these with pharmaceutical examples: e.g., asymmetric hydrogenation in the synthesis of L-DOPA (Parkinson’s drug), Sharpless epoxidation in making chiral epoxides for drugs like reboxetine, chiral auxiliaries in antibiotic synthesis, etc. Reaction pathway diagrams will be used to visualize how starting materials are converted to chiral products with control of stereochemistry.
Stereoselective vs. Stereospecific – Definitions
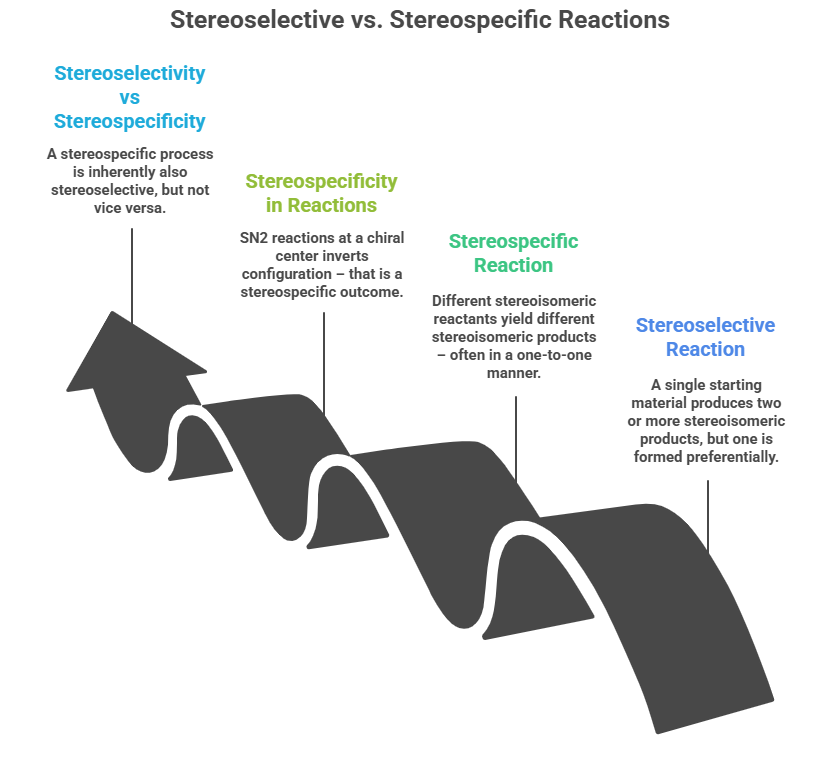
As defined by IUPAC, a stereoselective reaction is one where a single starting material can produce two or more stereoisomeric products, but one is formed preferentially. For instance, in an asymmetric hydrogenation of a prochiral alkene, both R- and S- hydrogenated products are possible, but a chiral catalyst might produce 95% R and 5% S – that’s stereoselective (enantioselective). The degree of stereoselectivity is often quantified by enantiomeric excess (ee) or diastereomeric excess (de).
In contrast, a stereospecific reaction implies that if you start from different stereoisomeric reactants, the outcomes are different stereoisomeric products – often in a one-to-one manner. A classic example: the bromination of cis-2-butene vs trans-2-butene. Cis gives meso-2,3-dibromobutane, trans gives racemic 2,3-dibromobutane. The mechanism (bromonium ion opening) is such that the stereochemistry of the starting alkene specifically dictates the product’s stereochemistry – thus it’s stereospecific. Another example is SN2 reactions: an SN2 at a chiral center inverts configuration – that is a stereospecific outcome (100% inversion if conditions are purely SN2). In drug synthesis, some reactions are stereospecific (e.g., certain concerted cycloadditions following orbital symmetry rules, or enzymatic reactions that act on one enantiomer in a pair). However, when we deliberately introduce chirality, we often rely on stereoselective approaches (not every reaction can be stereospecific, especially if starting from achiral substrates). A stereospecific process is inherently also stereoselective (if there’s only one outcome per stereochemistry, it’s selective by definition), but not vice versa.
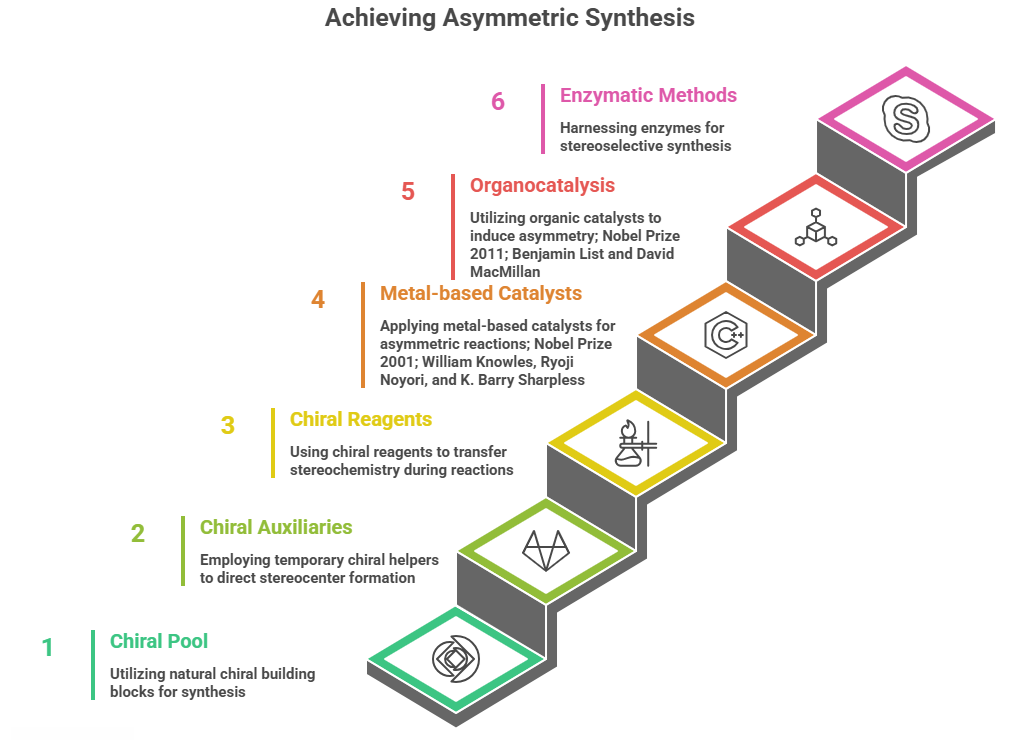
Strategies for Asymmetric Synthesis
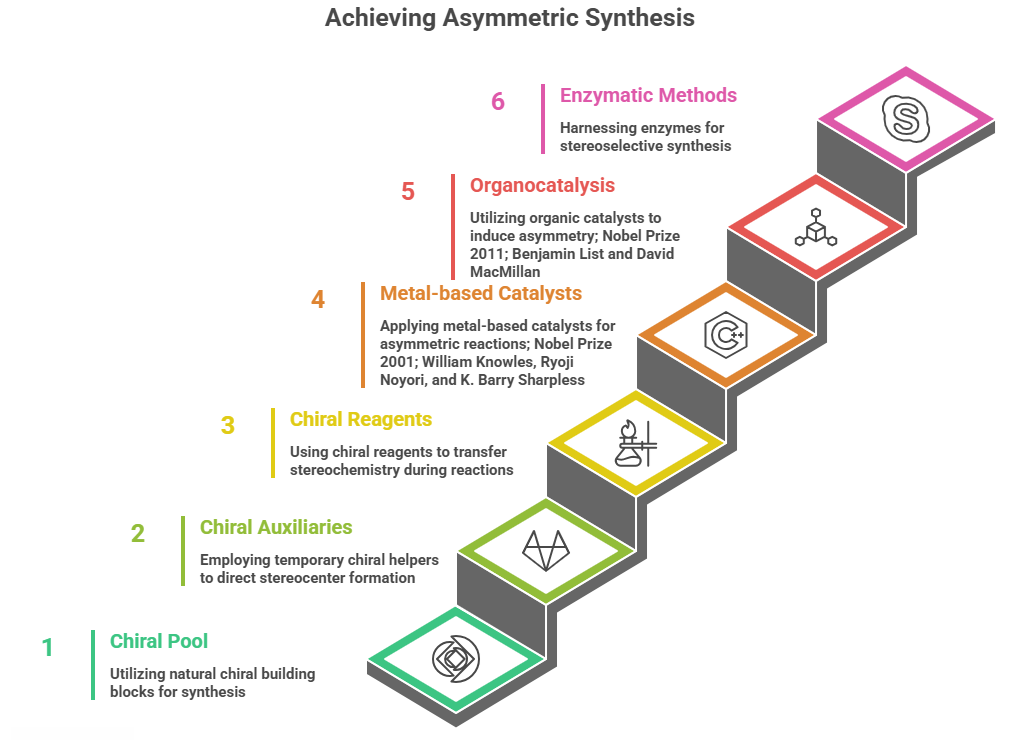
- Chiral Pool: This refers to starting from readily available enantiomerically pure building blocks obtained from nature (the “pool” of chirality in natural molecules). Many natural substances like amino acids, sugars, terpenes, and alkaloids are single enantiomers. These can be used as starting materials or key intermediates, carrying their chirality into the target. For example, L-menthol, L-tartaric acid, D-glucose, and L-amino acids have been widely used as chiral sources. A simple case: the synthesis of the anti-Parkinson’s drug L-DOPA initially was accomplished by resolving racemic D,L-DOPA. Later, Monsanto developed an asymmetric hydrogenation route using a chiral Rhodium catalyst (Knowles’ catalyst with a DIPAMP ligand) that hydrogenated an alkene precursor to yield L-DOPA in high enantiomeric purity. But to make the precursor, they started from an amino acid derivative (which has built-in chirality). That combination of chiral pool (the amino acid template) and chiral catalyst allowed an efficient stereoselective synthesis. The use of amino acids or hydroxy acids from nature has been key in many drug syntheses – it saves the trouble of creating chirality from scratch since nature already did. For instance, penicillin derivatives often start from L-cysteine or L-valine building blocks to ensure the correct stereochemistry in the β-lactam core.
- Chiral Auxiliaries: A chiral auxiliary is a temporary stereochemical helper that is attached to a substrate to direct the formation of a new stereocenter, then later removed (ideally recycled). One famous system is the Evans oxazolidinone auxiliary. Introduced by David Evans, these auxiliaries (derived from readily available chiral acids like valine) are attached to form an N-acyl oxazolidinone on a substrate. Then reactions such as enolate alkylations or aldol reactions proceed with high diastereoselectivity under the influence of the chiral auxiliary. Finally, the auxiliary can be cleaved off, yielding the desired product with a stereocenter and regenerating the auxiliary. For example, an Evans auxiliary could be used to synthesize a chiral intermediate of a drug like taxol (though taxol’s full synthesis is extremely complex, early steps used auxiliary-based aldol reactions to set multiple stereocenters). Another auxiliary: Oppolzer’s sultam (camphorsultam) has been used to control asymmetric Diels-Alder reactions. Case: In the synthesis of the anticancer agent epothilone, a chiral auxiliary guided a key cycloaddition to favor one diastereomer. Auxiliaries have been crucial in academic syntheses, but in industrial manufacturing, they add extra steps (attachment and removal), so their use is balanced against the cost of inefficiency. Nonetheless, for complex molecules where catalytic methods fail, auxiliaries remain a powerful tool.
- Chiral Reagents: These are reagents that themselves are chiral and transfer stereochemistry during the reaction. A classic example is the Brown asymmetric hydroboration – use of chiral borane reagents like Alpine-Borane (derived from α-pinene, a chiral terpene) to hydroborate alkenes, yielding chiral organoboranes that can be oxidized to alcohols with high enantioselectivity. H.C. Brown used reagents like diisopinocampheylborane to synthesize chiral alcohols. Another example is the CBS reduction (Corey–Bakshi–Shibata reagent) for asymmetric reduction of ketones to secondary alcohols, using a chiral boron hydride reagent with an oxazaborolidine catalyst (derived from proline). That’s more a catalyst actually, which segues to our next category. But one can consider Mosher’s acid chloride as a chiral derivatizing agent – not to create chirality, but to measure it (as used in NMR analysis). However, as reagents: Noyori’s chiral BINAP-Ru hydride is a catalyst, but if we speak of reagents consumed in stoichiometric amounts: Sharpless’ diethyl tartrate in epoxidation can be seen as a stoichiometric chiral modifier (though it acts catalytically with Ti).
- Chiral Catalysts: This is arguably the most elegant approach – using a small amount of a chiral catalyst to induce asymmetry in a reaction, meaning the catalyst’s chirality biases the formation of one enantiomer over the other in the product. These can be subdivided into metal-based catalysts and organocatalysts (metal-free).
- Metal-based: Many Nobel Prizes here – William Knowles and Ryoji Noyori (2001 Nobel) for asymmetric hydrogenation and asymmetric transfer hydrogenation; K. Barry Sharpless (2001 Nobel) for asymmetric epoxidation and dihydroxylation. Examples: Knowles’ Rh-DIPAMP catalyzed hydrogenation to make L-DOPA with >95% ee was a landmark (first industrial asymmetric catalyst for a drug). Another case: Asymmetric hydrogenation is now widely used to make chiral centers in drugs like the HIV protease inhibitor amantadine derivative (though amantadine itself is achiral) – better example: Sitagliptin (antidiabetic) manufacturing was optimized to use an asymmetric hydrogenation with a Ru-BINAP catalyst, replacing a previous resolution step, improving efficiency and reducing waste (Merck’s breakthrough with a Jack Halpern award). Sharpless epoxidation: This reaction (Ti(OiPr)<sub>4</sub> + diethyl tartrate + t-butyl hydroperoxide) epoxidizes allylic alcohols enantioselectively. It was used, for example, in the synthesis of the drug Reboxetine (an antidepressant). Reboxetine has two stereocenters; one approach to make it involved a Sharpless epoxidation of an allylic alcohol to set those centers in a precursor. Specifically, the (2R,3R)-epoxy alcohol intermediate was obtained by Sharpless epoxidation of cinnamyl alcohol (allylic alcohol) using (+)-diethyl tartrate, leading into a sequence that yielded (R,R)-reboxetine. (Reboxetine is marketed as a racemate actually, but synthetic routes exist to make the enantiomers.) Another Sharpless method: Sharpless asymmetric dihydroxylation (using OsO4 with chiral quinine ligands) is used in some drug syntheses for vicinal diols (e.g., in making certain statin side-chains). Jacobsen’s epoxide hydrolysis (kinetic resolution) – an alternative method to get chiral epoxides or diols (though it’s a resolution, not a direct asymmetric synthesis – more in Part 6).
- Organocatalysis: The use of purely organic, small-molecule catalysts to induce asymmetry has boomed in the 21st century, earning MacMillan and List the 2021 Nobel Prize. An example is the proline-catalyzed asymmetric aldol reaction (Hajos–Parrish–Eder–Sauer–Wiechert reaction) known since the 1970s for making steroid precursors – proline, a chiral amino acid, catalyzes intramolecular aldol reactions stereoselectively. More recent organocatalysts include MacMillan’s imidazolidinone catalysts for asymmetric Diels–Alder and Michael reactions, and Jørgensen’s catalysts for asymmetric epoxidation (e.g., Jørgensen’s amino-thiourea catalysts for epoxidation of enals). In pharma, organocatalysis has been applied to, for example, the synthesis of the antiviral oseltamivir (Tamiflu) – one step in a route uses a proline-catalyzed aldol reaction to set a stereocenter. Another example: the anti-inflammatory naproxen can be synthesized by an organocatalytic enantioselective reaction (though in practice it’s often resolved or semi-synthesized). The advantage of organocatalysts is they often avoid metals (which is greener and avoids metal contamination concerns in APIs). They can sometimes be used in combination (sequentially or cooperatively) to construct complex molecules.
- Enzymatic and Chemoenzymatic Methods: Enzymes are nature’s chiral catalysts, and they are increasingly harnessed in drug synthesis for their high stereoselectivity and mild conditions. For example, lipases can asymmetrically hydrolyze one enantiomer in a ester (kinetic resolution) or desymmetrize prochiral diesters to monoesters. A notable case: the manufacture of the antibiotic sitagliptin (Januvia) at one point involved a transaminase enzyme to set a chiral amine stereocenter, providing near-complete conversion to the desired enantiomer (Merck later improved it with an enzyme, replacing a rhodium catalyst method). Epoxide hydrolases (as mentioned in the BASF snippet can selectively open one enantiomer of a racemic epoxide (kinetic resolution) to give diols with high enantiomeric purity, or if done on a meso-epoxide can desymmetrize it into a chiral diol. Biocatalytic routes are often used for manufacturing chiral building blocks: e.g., using dehydrogenase enzymes to reduce ketones to chiral alcohols (with cofactor recycling), or ammonia lyases to produce L-amino acids, etc. Many pharmaceutical building blocks (like chiral amines or alcohols) are now made at scale with enzymes (engineering of enzymes via directed evolution, like Frances Arnold’s work, Nobel 2018). For instance, the chiral glycidol (epoxy alcohol) used in some drug syntheses can be made via enzymatic routes or resolution.
💡 “Explore asymmetric synthesis in detail through our blog series: #Asymmetric_Synthesis”
Pharmaceutical Examples of Asymmetric Synthesis
- L-DOPA via Asymmetric Hydrogenation: Mentioned above, the first industrial asymmetric catalytic reaction in pharma. Knowles’ rhodium catalyst gave >95% e.e., winning him a share of the Nobel. This was stereoselective (the catalyst favored L over D product strongly) and stereospecific in that the olefin geometry correlated to product stereochemistry (but here substrate was achiral, so we say stereoselective).
- Beta-blocker side chain: The (R)-alcohol in beta-blockers like propranolol can be synthesized by Noyori’s asymmetric transfer hydrogenation of acetophenone derivatives using Ru(II) with a chiral diamine ligand, obtaining high yield of one enantiomer.
- ACE inhibitors (e.g., Enalapril): Enalapril has a chiral center. One route to enalaprilat (active acid form) uses an enzymatic desymmetrization of an anhydride by a lipase to introduce chirality, followed by chemical steps to assemble the molecule.
- Atorvastatin (Lipitor): Synthesis of its side-chain (a dihydroxyheptanoic acid) initially involved diastereoselective aldol reactions (with Evans auxiliary) to set the two stereocenters. Later processes used chiral catalysis or biocatalysis to avoid auxiliary.
- Antibiotics: The glycosylated natural product drugs (like erythromycin derivatives) rely on fermentation or semisynthesis because total synthesis would involve too many stereocenters. However, simpler synthetic antibiotics (like chloramphenicol early on was resolved, or linezolid which is made by stereoselective synthesis) show that more chiral drugs can now be synthesized rather than isolated.

Green Chemistry and Efficiency
Asymmetric synthesis strategies also tie into “green chemistry”. Stereoselective methods, especially catalytic ones, avoid the waste of resolving racemates (which at best give 50% yield of desired enantiomer unless the other enantiomer can be racemized and recycled – a process called dynamic kinetic resolution). Many pharma companies strive to implement asymmetric catalysis or biocatalysis early in synthesis to maximize yield of the correct stereoisomer and minimize extra steps. This points to a future where more drug syntheses will employ enzyme catalysts or well-designed organometallic/organocatalysts for stereocontrol.
Asymmetric synthesis is going greener—driven by the 12 principles of green chemistry. Read more @ <https://chiralpedia.com/blog/the-future-asymmetric-synthesis-trends-and-innovations/>.
Stereospecific Reactions in Drug Synthesis
While most efforts are about achieving stereoselectivity, some reactions are inherently stereospecific and used in drug synthesis. For example, if one has an E- vs Z- intermediate, one might exploit that in different routes (like making only the E isomer because only that leads to the correct stereochemistry in a cyclization). Or using an SN2 displacement on a known chiral center (from natural chiral pool) to invert it if needed. A trivial but common stereospecific step: using inversion to get the desired enantiomer from the undesired one (via a one-step invert of configuration, e.g., Mitsunobu reaction to invert a secondary alcohol). If one enantiomer of a secondary alcohol is easier to get, chemists might Mitsunobu-invert it to the other if needed (stereospecific 100% inversion if conditions ideal). For instance, early syntheses of some prostaglandins did such flips.
Summary (Part 5)
– Stereoselective synthesis: Reactions or sequences that favor the formation of one stereoisomer (enantiomer or diastereomer) over others. Key measure: enantiomeric excess (ee) or diastereomeric ratio. Essential for making single-enantiomer drugs efficiently.
– Stereospecific reactions: Those in which the stereochemical outcome is strictly dictated by the stereochemistry of the reactant(s). In practice, stereospecificity is a property of certain mechanisms (SN2, concerted additions, etc.) and can be harnessed (e.g., use SN2 to invert configuration in a known chiral center).
– Chiral pool: Utilizing naturally occurring chirality (amino acids, sugars, terpenes). Advantage: often enantiomerically pure and inexpensive on large scale (e.g., L-amino acids produced via fermentation). Many older syntheses rely on these (e.g., L-aspartic acid in making anti-asthma drugs, L-tartaric acid for chiral pool in many syntheses, etc.).
– Chiral auxiliaries: Temporary attachments that induce diastereoselectivity in a key step. Examples: Evans’ oxazolidinones in acylation/aldol/alkylation (widely used in the 80s/90s for making chiral intermediates for drugs like renin inhibitors, statins, etc.), Oppolzer’s camphorsultam (for Diels-Alder control), etc. Auxiliaries require extra steps but often give very high stereoselectivities (diastereomeric ratios).
– Chiral reagents: Reagents that themselves are chiral and transfer that chirality in reaction. E.g., Alpine borane for hydroboration, or chiral diazomethane equivalents for Simmons-Smith cyclopropanations, etc. Many have niche uses.
– Chiral catalysts: The modern workhorse: asymmetric catalysis (metal complexes like Rh-, Ru-, Pd-, Ir- catalysts with chiral ligands, or small organic catalysts). They enable lower catalyst loadings and avoid stoichiometric chiral materials, thus more economical and green. Examples:
– Rh–BINAP or Ru–BINAP for asymmetric hydrogenations (pharma processes for amino acids, chiral amines, alcohols – e.g., Ru–BINAP for (R)-1,2-propanediol in levofloxacin synthesis).
– Sharpless epoxidation/dihydroxylation (Ti-Tartrate or OsO4-quinine systems) – used in making complex drug intermediates (e.g., certain chiral building blocks for beta-blockers, leukotriene antagonists).
– Organocatalysis – proline-catalyzed aldols (used in steroid syntheses), MacMillan catalysts in alkaloid syntheses, etc. Industry uptake is growing as some organocatalysts become more robust and available at scale.
– Biocatalysis – enzymatic resolutions or asymmetric syntheses (transaminases to make chiral amines, reductases for alcohols, hydrolases for esters). Many pharmaceutical companies now engineer enzymes for specific steps (e.g., the Atorvastatin side-chain synthesis uses a dehydrogenase cascade).
– Examples in pharma: L-DOPA by Rh-catalyzed hydrogenation; (S)-metolachlor (herbicide, not a drug but a large-scale example) by chiral iridium catalyst hydrogenation; Sitagliptin by transaminase enzyme; Escitalopram (antidepressant) by resolution historically, but newer routes use asymmetric step; synthesis of antiviral oseltamivir employing organocatalytic Diels-Alder; etc. Each example demonstrates a principle (resolution vs asymmetric synthesis vs chiral pool).
– Stereospecific vs. selective in context: Many enzymatic reactions are stereospecific – e.g., an enzyme might only convert L-substrate and leave D (hence a kinetic resolution). A synthetic chemist designs sequences to maximize stereoselectivity and uses stereospecific steps to maintain or invert stereochemistry as needed.
Impact on drug development
Suggested Reading
IUPAC Gold Book for definitions: stereoselective <https://goldbook.iupac.org/terms/view/S05991#:~:text=The%20preferential%20formation%20in%20a,expressed%20by%20the%20diastereoisomer%20excess>, stereospecific <https://goldbook.iupac.org/terms/view/S05994#:~:text=1,stereospecific%20process%2C%20although%20the%20analogous>. (Useful to ensure correct usage of these terms with examples provided in the definitions.)
Modern Synthetic Methods: Asymmetric Synthesis – The Essentials (eds. Christmann & Bräse, 2007). A concise reference that covers auxiliary-based methods, catalysts, etc., with examples from pharma.
Nobel Lectures 2001: Sharpless, Noyori, and Knowles – published in Angew. Chem. Int. Ed. (2002). They describe the development of their catalytic methods and mention industrial applications like L-DOPA and others.
https://www.nobelprize.org/prizes/chemistry/2001/popular-information
https://www.nobelprize.org/prizes/chemistry/2021/popular-information
Review on Biocatalysis in pharma: Pollard, D., & Woodley, J. (2007). “Biocatalysis for pharmaceutical intermediates: The future is now.” Trends in Biotechnology, 25(2), 66–73. (Outlines how enzymes are used for chiral synthesis with case studies including statins, etc.)
Org. Process Research & Dev. articles on specific drugs: e.g., Sitagliptin manufacture (2010, Vol. 14, p. 38) – describes switching from Rh catalyst to transaminase enzyme; Atorvastatin synthesis improvements (2008, Vol. 12, p. 81) – use of asymmetric hydrogenation.
