“Why one enantiomer heals while the other may harm – the pharmacology of chirality”
Introduction
Chirality doesn’t just influence drug properties in theory – it has very real consequences in pharmacology. In this section, we explore how stereochemistry affects drug action at multiple levels: pharmacodynamics (drug-receptor interactions) and pharmacokinetics (absorption, distribution, metabolism, excretion). We will define terms like eutomer (the more active enantiomer) and distomer (the less active one) and introduce the concept of the eudismic ratio (the ratio of activities of enantiomers). Through famous case studies – thalidomide, ibuprofen, omeprazole, and others – we will see examples of enantioselectivity in therapeutic and toxic effects. This part underlines why, for many drugs, it is not sufficient to develop a racemate without understanding each enantiomer’s behavior.
Pharmacodynamics – Chirality in Drug-Receptor Interactions
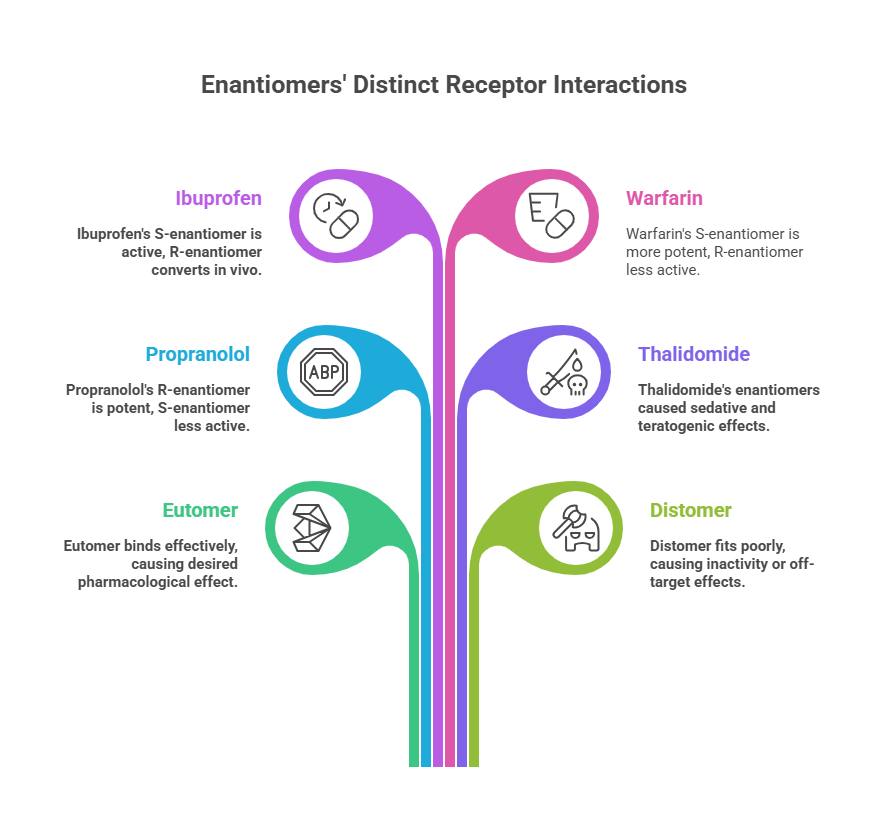
Most biological targets (receptors, enzymes, ion channels, DNA) are chiral environments. Imagine a receptor as a glove: it can distinguish between a left hand and a right hand. Thus, enantiomers of a chiral drug often have different affinities or intrinsic activities at the target site. One enantiomer may bind snugly to the receptor’s binding pocket (like the correct hand in a glove) – this is typically the eutomer, responsible for the desired pharmacological effect. The mirror-image drug may fit poorly or differently – distomer; it could be inactive, or sometimes it binds to a different site or receptor altogether, potentially causing off-target effects. [Read more @ <https://chiralpedia.com/blog/chiral-pharmacology-the-mirror-image-of-drug-development/>.
Case Study 1: Propranolol
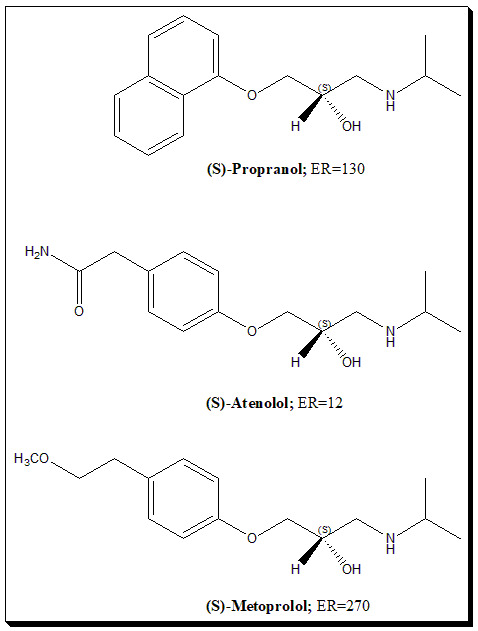
For example, propranolol, a non-selective β-blocker: the l-(–)-propranolol enantiomer (also known as S-propranolol) is a potent β-adrenergic antagonist, while d-(+)-propranolol (R-propranolol) is ~100-fold less active at β-receptors. This is because the binding pocket of the β-receptor “sees” the two enantiomers as different shapes. In clinical use, propranolol was introduced as a racemate, but essentially all the β-blocking activity comes from the l-enantiomer. The other enantiomer is considered mostly inert in terms of the primary action (though it might contribute to side effects or have different pharmacokinetics).
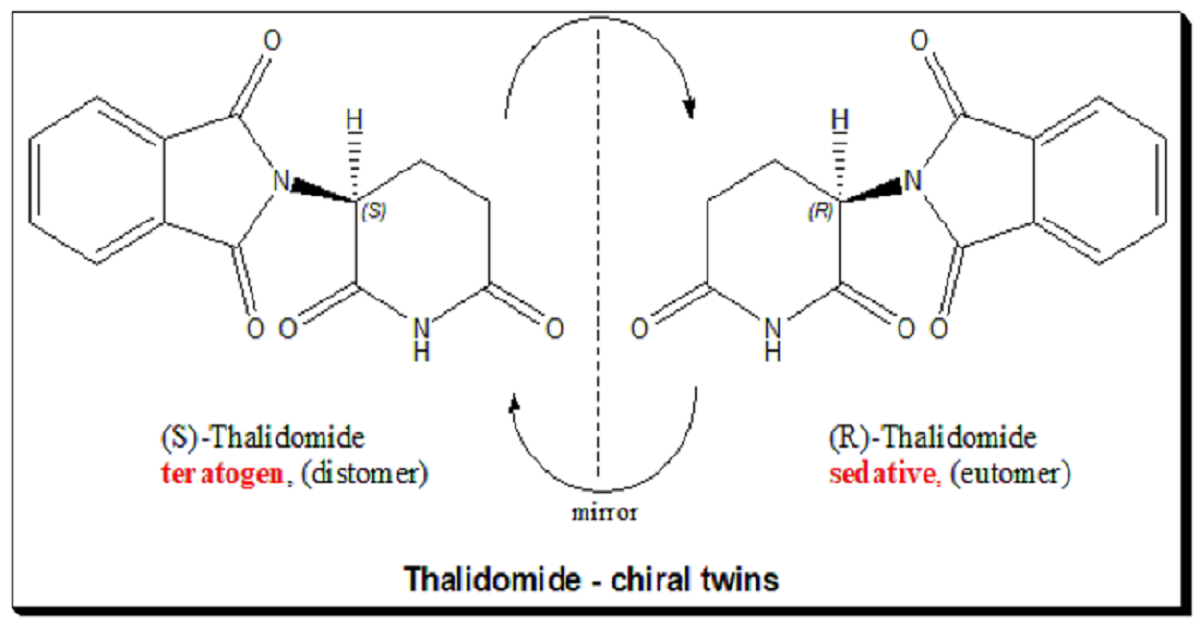
Case Study 1: Thalidomide
Another striking example: Thalidomide. This is a textbook case where chirality literally spelled the difference between a drug and a poison. However, a complicating factor is that thalidomide racemizes in vivo (interconverts between enantiomers), so even administering a single enantiomer wouldn’t have completely averted tragedy – an important cautionary note we’ll revisit in Part 6. Nonetheless, this case alerted the world to the importance of evaluating each enantiomer of chiral drugs separately. [For an in-depth look, explore the full blog 👉 <https://chiralpedia.com/blog/thalidomide-tragedy-the-story-and-lessons/>.
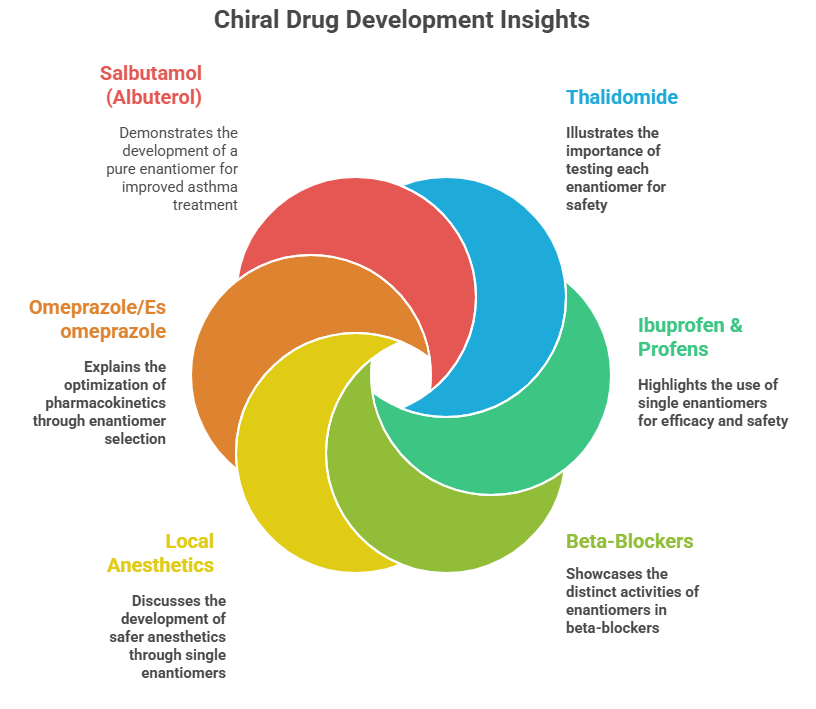
Case Study 3: Ibuprofen
A classic case in analgesics is ibuprofen. Ibuprofen has a chiral center in the propionic acid side chain. The marketed product for decades was racemic ibuprofen. It was known that the S-(+)-ibuprofen is the active form that inhibits cyclooxygenase (COX) and thus inflammation/pain. The R-(–)-ibuprofen is much less effective at COX (eudismic ratio for COX inhibition is significant). Interestingly, humans (and many animals) have an enzyme (an isomerase in the liver) that slowly converts R-ibuprofen to S-ibuprofen. In vivo studies showed that about 60% of R-ibuprofen could be converted to the active S-form in some populations, though in others (like certain ethnic groups) the conversion was far less (only ~25% in Chinese populations). This partially mitigates the disadvantage of selling ibuprofen as a racemate – some of the “less active” enantiomer is turned into the active one in the body. Nevertheless, recognizing the advantage of a single-enantiomer product, a pure S-(+)-ibuprofen (called dexibuprofen) was later developed and marketed in some countries, offering potentially faster onset or efficacy at lower doses, though clinical differences are modest. This highlights how metabolism and pharmacodynamics together must be considered: the seemingly inactive enantiomer of ibuprofen isn’t completely wasteful because it can activate via chiral inversion in vivo, but that depends on individual metabolism.
Case Study 4: Warfarin
Warfarin, an anticoagulant. Warfarin’s enantiomers are both active as anticoagulants (they inhibit vitamin K epoxide reductase, VKOR, which is involved in clotting factor maturation). However, S-warfarin is about 3–5 times more potent than R-warfarin in terms of anticoagulant effect. They are metabolized by different cytochrome P450 enzymes: S-warfarin is primarily metabolized by CYP2C9, whereas R-warfarin by CYP3A4 and others. As a result, drug interactions or genetic polymorphisms in CYP2C9 affect S-warfarin’s clearance more – making the pharmacokinetics of the racemate a composite of two different half-lives. Clinically, warfarin is still used as the racemate (Coumadin), but dosing and monitoring (INR checks) account for the net effect. This case shows that even when both enantiomers share the same qualitative activity (both anticoagulant), one might be considered the main contributor while the other is weaker and hangs around longer. Indeed, this can cause complications: differences in enantiomer metabolism mean that the enantiomeric composition in plasma can shift over time or with interactions.
Eudismic Ratio and Enantioselectivity
The eudismic ratio (ER) is a concept that quantifies how big the activity difference is between two enantiomers. It can be defined, for instance, as the ratio of EC50 or IC50 of the distomer to the eutomer (so a larger number indicates a bigger difference). A high eudismic ratio means one enantiomer is much more potent than the other. For example, if the (S)-enantiomer of a hypothetical drug has an EC50 of 1 nM at a receptor, and the (R)-enantiomer’s EC50 is 100 nM, the eudismic ratio is 100. Such information guides drug development decisions: if one enantiomer is overwhelmingly more active, there is a strong argument to develop only that enantiomer as the drug (to minimize dosing and side effects from the inert or harmful partner). If both enantiomers have similar activity (ratio near 1), a racemate might suffice.
Reflecting the degree of stereoselectivity
Enantioselective Pharmacokinetics
Beyond receptor binding, chirality can influence all ADME aspects:
– Absorption: Sometimes one enantiomer is absorbed faster or to a greater extent. For example, L-amphetamine is absorbed more readily than D-amphetamine from the gut. The reasons can be complex (differential transporter affinity or first-pass metabolism differences).
– Distribution: Plasma protein binding or tissue distribution can differ. One enantiomer might fit better into plasma albumin’s binding site, altering free drug levels.
– Metabolism: As seen with warfarin, different enantiomers often have different metabolic routes and rates because enzymes (being chiral proteins) may preferentially recognize one over the other. Another example: omeprazole (mentioned earlier). Omeprazole is a proton pump inhibitor marketed originally as a racemate (Prilosec). Its S-enantiomer, esomeprazole, was later marketed. What was the advantage? It turns out S-omeprazole is metabolized more slowly than R-omeprazole in humans – specifically, the S-enantiomer has lower clearance and less inter-individual variability. This leads to higher and more consistent plasma levels of the active drug, for a given dose, when using pure S compared to the racemate. In practical terms, 20 mg of esomeprazole (S-only) can achieve acid suppression comparable to 40 mg of racemic omeprazole, and with less variability between patients. The reason is enantioselective metabolism: R-omeprazole undergoes faster first-pass metabolism via CYP2C19, whereas S-omeprazole is metabolized a bit slower and more by CYP3A4; plus some individuals are poor metabolizers for R due to CYP2C19 genetics. By using the single S-enantiomer (and doubling the dose relative to one enantiomer’s contribution in racemate), AstraZeneca could both extend the patent life (a so-called “racemic switch” strategy and claim a pharmacokinetic benefit. This illustrates how one enantiomer can be preferred not necessarily because the other is inactive, but because the other is less predictable or efficient in the body.
– Excretion: If a drug’s excretion involves chiral interactions (like active transport in kidneys), enantiomers might be excreted at different rates.

Case Studies
- Thalidomide: As mentioned, R-thalidomide (as commonly designated in literature) was intended as a sedative, S-thalidomide was teratogenic[4]. This taught drug developers to test each enantiomer of a chiral drug for safety. Interestingly, thalidomide’s teratogenic mechanism was later linked to its binding to a protein (cereblon) involved in limb development, and that binding is stereoselective. Modern analogs of thalidomide (lenalidomide, pomalidomide) are used to treat cancers, and they too are chiral and are used as single enantiomers – with much caution due to the same target but in a controlled context (and obviously contraindicated in pregnancy).
- Ibuprofen & Profens: Ibuprofen (Advil) we covered: only S is active, R is converted partly. Similarly, other “profens” (naproxen, ketoprofen, flurbiprofen, etc.) have one active enantiomer. Notably, naproxen is sold as a single enantiomer (the S-naproxen, as the sodium salt Aleve). The R-naproxen was found to cause liver toxicity in test animals and is not used, plus S-naproxen doesn’t racemize. Naproxen’s case is a straightforward reason to go single enantiomer from the start.
- Beta-Blockers: Many beta-blockers are chiral (propranolol, alprenolol, atenolol, metoprolol, etc.). Usually, the S-enantiomer carries the β-blocking activity (because they were optimized that way). Early beta-blockers were used as racemates (e.g., propranolol). Some later ones like levobunolol (for glaucoma) were introduced as single enantiomers (“levo” indicating the active one). Another twist is sotalol – interesting for having two very different activities: the d-sotalol enantiomer is a Class III antiarrhythmic (potassium channel blocker), while l-sotalol is a beta blocker (Class II). Racemic sotalol has both actions and is used to treat arrhythmias, but d-sotalol alone was investigated to avoid beta-blockade and unfortunately found to increase mortality in some patients, so it was not pursued alone. Here, each enantiomer had a distinct pharmacology – a rare but striking case of “functional enantiomers” with different therapeutic profiles (they even fell into different drug classes).
- Local Anesthetics: Bupivacaine is an anesthetic that is chiral. Both enantiomers cause nerve block, but the R-(+)-bupivacaine was associated with more cardiotoxicity. A single S-(–)-enantiomer formulation, levobupivacaine, was developed to provide a potentially safer anesthetic with similar efficacy but lower cardiac risk. Similarly, etomidate (an IV anesthetic) – one enantiomer is a potent hypnotic, the other is much less so.
- Omeprazole/Esomeprazole: Already discussed as an example of optimizing PK by choosing one enantiomer. Esomeprazole (S-omeprazole) became a blockbuster on its own, partly as a lifecycle management strategy (since omeprazole’s patent was expiring), but also marketed with the claim of more consistent acid control due to less inter-patient metabolic variation.
- Salbutamol (Albuterol): This common bronchodilator is a racemate. The bronchodilating activity resides mainly in R-(–)-salbutamol. The S-(+)-enantiomer was long thought inert, but some studies indicated it might actually have pro-inflammatory or bronchoconstrictive effects. This led to the development of levalbuterol (Xopenex), the pure R-enantiomer, as a potentially superior asthma treatment with fewer side effects. Clinically, levalbuterol produces similar bronchodilation at half the dose of racemic albuterol, supporting that essentially only R is active. The benefits in terms of side effect reduction are modest, but it’s another example of a chiral switch in therapy.
Implications for Drug Development: Stereochemistry and the Future of Pharma
All these examples underscore why regulatory agencies require thorough evaluation of stereochemistry early in development. An overview of the above case-studies presented visually below.
Drug regulators pay close attention to stereochemistry because each “hand” of a molecule can act very differently in the body. The FDA advises testing enantiomers separately to see which one brings the benefit and which might cause harm. Sometimes both are fine, but other times only one is worth keeping. This is why many drugs have been “switched” from racemic mixtures to single-enantiomer versions, like esomeprazole or levocetirizine. The catch? Single-enantiomer drugs aren’t always better — it depends on the case. Still, today’s drug discovery trend leans toward developing pure stereoisomers whenever possible, to boost effectiveness and reduce unwanted surprises.
Summary (Part 4)
– Enantiomers in pharmacodynamics: Often exhibit different potency or efficacy at targets (one fits receptors better – the eutomer). This can lead to one enantiomer being responsible for most of a drug’s therapeutic effect. Examples: S-metoprolol (active β-blocker) vs R-metoprolol; S-(+)-ibuprofen (active COX inhibitor) vs R-ibuprofen (inactive until converted).
– Eutomer/Distomer and Eudismic Ratio: Terms to describe which enantiomer is more active and how large the difference is. Large differences favor development of single-enantiomer drugs for better efficacy and safety profile.
– Enantiomers in pharmacokinetics: Can be absorbed, distributed, metabolized, or excreted at different rates. One enantiomer may have a longer half-life or be processed by a different enzyme (leading to interactions or variability). E.g., R-warfarin vs S-warfarin metabolism by different CYPs, R-omeprazole vs S-omeprazole metabolism.
– Case studies:
– Thalidomide: One enantiomer teratogen, one sedative – taught the world to test each enantiomer; but in vivo racemization complicates matters.
– Ibuprofen: Only S is active, R converts to S partly; racemate still used, but S-enantiomer introduced in some markets.
– Omeprazole/Esomeprazole: S-enantiomer chosen for improved PK consistency, an example of a racemic switch (and patent extension).
– Beta-blockers: Typically only one enantiomer has β-blocking activity (the other is essentially filler or might cause side effects).
– Sotalol: Unusual case where enantiomers have entirely different pharmacological profiles (one beta-blocker, one antiarrhythmic).
– Local anesthetics and others: Often one enantioform is safer (less toxic side effects) – e.g., levobupivacaine.
– Regulatory perspective: Developers must justify choosing a racemate over a single enantiomer if one enantiomer is superior. Many new drugs are developed as pure enantiomers unless a racemate is unavoidable or both enantiomers contribute usefully. Guidelines require studying the pharmacology and toxicology of individual enantiomers early on.
– Therapeutic outcomes: The use of pure enantiomers can sometimes reduce side effects (no distomer to cause off-target effects) and simplify pharmacokinetics, but not always a dramatic difference in efficacy as meta-analyses have shown. Each case is unique; for example, levocetirizine (Xyzal) vs cetirizine (Zyrtec) – levocetirizine is the active enantiomer of cetirizine and provides the same antihistamine effect at half the dose, with similar clinical outcomes, mainly offering a marketing advantage of “less drug needed” and possibly slightly less sedation.
Suggested Reading
Caldwell, J. (1995). “Importance of stereochemistry in drug action.” Eur. J. Clin. Pharmacol., 48, 5–8. (Discusses multiple examples of enantioselective drug action and the concept of eudismic ratio in pharmacology.)
FDA Guidance for Industry (1992). Sections II and III[57][58]. (Policy statements on considering single enantiomers vs racemates in development, with reasoning and examples.)
The Significance of Chirality in Drug Design and Development” – M. E. Ariëns, Medicinal Research Reviews, 7(5), 1987. (A classic paper by Ariëns coining terms like eutomer/distomer and discussing many case studies of chiral drugs.)
American Pharmaceutical Review Article[3][14] (2024) on chiral drug analysis – specifically the introductory sections that outline how enantiomers can have different pharmacologic or toxicologic profiles and mentions examples like ibuprofen’s conversion and meta-analysis findings on single vs racemic efficacy.
Medical chemistry textbooks (e.g., Foye’s Principles of Medicinal Chemistry, latest ed., chapter on stereochemistry in drug action) – provide numerous drug-specific anecdotes of stereochemical effects in pharmacology.
Patricia Van Arnum, Single-Enantiomer Drugs Drive Advances in Asymmetric Synthesis. Pharmaceutical Technology-04-02-2006
https://www.pharmtech.com/view/single-enantiomer-drugs-drive-advances-asymmetric-synthesis
