“From hit to lead to medicine – where stereochemistry shapes every stage of discovery”
Introduction
Stereochemistry is not only crucial in the final stages of drug production – it plays a significant role right from the discovery and lead optimization phases. In this part, we consider how medicinal chemists account for chirality when designing compounds and how structure-activity relationships (SAR) can depend on stereochemistry. We also look at how screening libraries incorporate stereochemical diversity, and how computational drug design deals with stereochemistry. Additionally, we outline the regulatory guidelines (ICH/FDA/EMA) that influence development decisions regarding chirality: from investigational new drug (IND) applications to new drug applications (NDA), requiring documentation of stereochemical composition and justification for racemate vs enantiomer development.
Structure-Activity Relationship (SAR) and Chirality
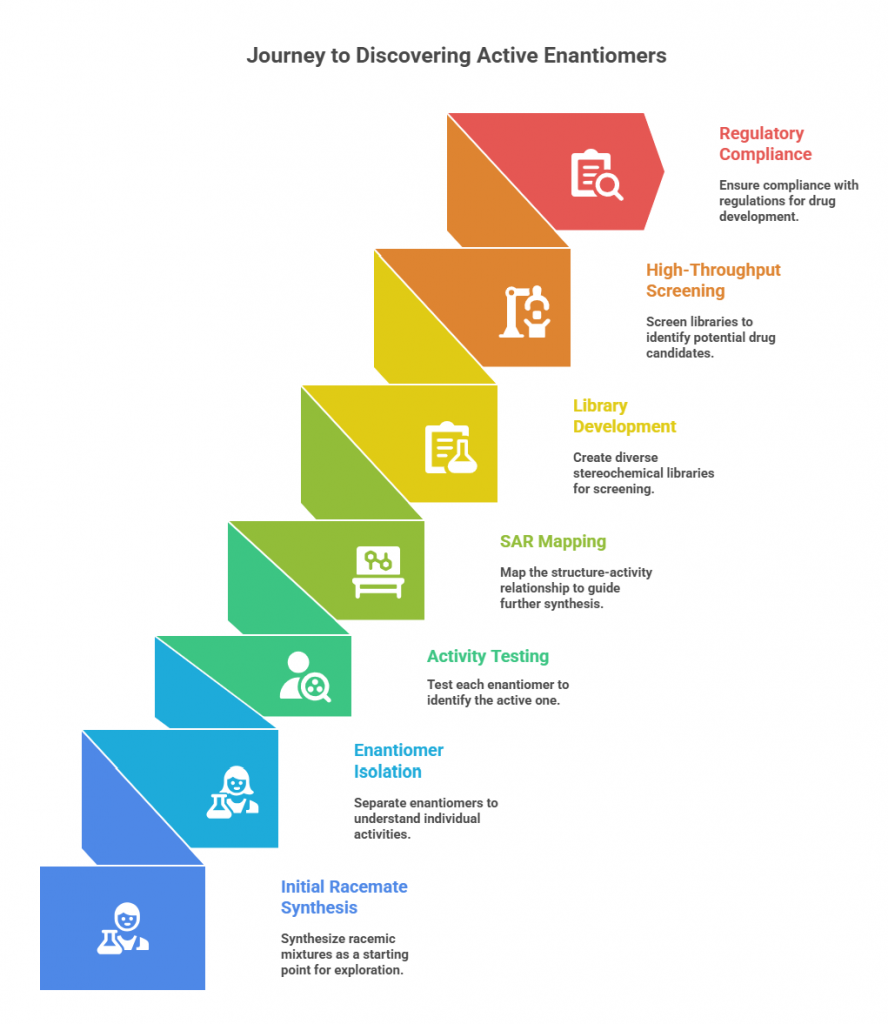
Medicinal chemistry is often about finding the right 3D arrangement of atoms that interacts optimally with a biological target. When a potential pharmacophore has one or more stereocenters, each stereoisomer can be considered a distinct molecule in SAR exploration. Often, initial synthetic schemes produce racemates, and these may show activity – but to truly understand SAR, chemists will try to isolate or resynthesize each enantiomer. A well-known pattern is that one enantiomer is the eutomer with significantly greater activity (higher receptor binding affinity, etc.) than the other. For example, in early β-blocker development, once it was recognized that the activity resided mostly in the l-enantiomers, medicinal chemists focused on synthesizing analogs and keeping (or testing) the configuration that gave activity.
Thus, medicinal chemists either (a) resolve racemic hits from a screen to see which enantiomer is active, or (b) design asymmetric syntheses to make specifically the stereochemistry believed to be needed. For instance, if a screening hit is a chiral molecule, they might use chiral chromatography to separate the enantiomers and test them individually. If one is completely inactive, resources can be concentrated on the active enantiomer series. If both have activity but different profiles, that might open investigation into dual-action drugs or separate development.
In some cases, both enantiomers of a lead series are synthesized and tested to map the “chiral SAR”. A famous example: propiconazole (a fungicide) – out of four stereoisomers, one had the best fungicidal activity and another had some different spectrum; by knowing that, chemists could refine which stereochemistry to keep in analogs. In pharmaceuticals, consider dopa analogs: early on, both D- and L-amino acid analogs were made, but only L is active in human enzymes (e.g., L-DOPA effective for Parkinson’s, D-DOPA not so). Knowledge of stereochemical SAR guides focus.
When a molecule has multiple stereocenters, the number of stereoisomers grows (2n). It’s impractical to test all if n is high, but often one starts from a natural product or known active which already has a certain stereochemistry. For synthetic projects, medicinal chemists sometimes make stereochemical libraries – for example, make both cis and trans versions of a ring junction, or both R and S at a key center, to see which orientation is optimal in binding. If one orientation is far superior, future analogs will maintain that.
Stereochemical Libraries and 3D Diversity
There’s been a realization that many historical screening libraries were biased toward flat aromatic compounds (achiral, low 3D complexity). In modern drug discovery, there’s emphasis on increasing Fsp3 (fraction of sp3 carbons) and introducing stereogenic centers to explore more 3D shape space. This is because more complex 3D structures can sometimes achieve higher target specificity or avoid P450 metabolism, etc. Companies have developed “3D-enriched libraries” which inherently include chiral molecules. Some of these libraries include compounds as racemic pairs initially (because synthesizing enantioenriched versions of thousands of library compounds can be resource-intensive). When a racemic hit is found, then resolution or resynthesis occurs as mentioned. Alternatively, some libraries deliberately include both enantiomers of chiral scaffolds as separate entries, to ensure screening doesn’t miss an active enantiomer.
For instance, Life Chemicals (in the snippet provided) offers 3D-shaped compound libraries and fragment libraries with stereochemical diversity. These sets have many chiral compounds; they might supply them as racemates or single enantiomers depending on feasibility. High-throughput screening (HTS) itself doesn’t inherently distinguish enantiomers unless they are separate entries. The blog excerpt notes the advantages and challenges: testing multiple enantiomers increases chances of finding activity, but preparing/purchasing both enantiomers doubles the number of compounds and cost, and you then have to separate or individually synthesize. The excerpt also notes regulatory compliance: even at screening stage, they consider the cost of making pure enantiomers (some screens may still use racemates and then follow up with chiral separation later).
High-Throughput Screening (HTS) considerations
– Opportunity: If racemates are screened, one might get a hit signal but not know which enantiomer (or if both contribute). This requires follow-up. HTS sensitivity can sometimes detect subtle differences if the racemate has partial activity (but typically you just get an averaged response). The blog says HTS can detect subtle differences in activity of enantiomers if both are tested and can quickly evaluate many compounds. – Challenge: Including both enantiomers of each library molecule doubles library size and cost. Some companies compromise by focusing on one enantiomer (say the one from natural pool or arbitrarily R) in the library, hoping if it’s active the enantiomeric pair is not needed. But that risks missing an active one if the chosen configuration was the wrong one. – Strategy: Sometimes, libraries are made as racemic mixtures for initial screening (which halves the number of screening entries). If a racemic well is a hit, in follow-up they resolve or resynthesize to get the pure enantiomers. This is a pragmatic approach but one must be cautious: if one enantiomer is an antagonist and the other an agonist, a racemate might show a null result and you’d miss it entirely – a scenario albeit rare but possible (one enantiomer could counteract the other’s effect). As a result, some screening groups prefer to test enantiomers separately when possible.
Computational Drug Design and Stereochemistry
In silico methods (docking, virtual screening) must account for stereochemistry of molecules. This means when you enumerate a virtual library, you should include all relevant stereoisomers of each structure (if they are stable and plausible) because different stereoisomers will have different 3D shapes and can bind differently to chiral protein targets. Many docking programs will automatically consider both enantiomers if not specified, or at least allow one to dock a specific enantiomer. Pharmacophore modeling similarly must consider chirality: a receptor may prefer a certain handedness. Some famous computational failures occurred when chirality was neglected – e.g., a model might overlay an S-enantiomer of one ligand with an R-enantiomer of another erroneously if chirality is not encoded, leading to a misleading hypothesis. Modern computational pipelines treat enantiomers as distinct structures (there is typically a flag to consider stereochemistry).
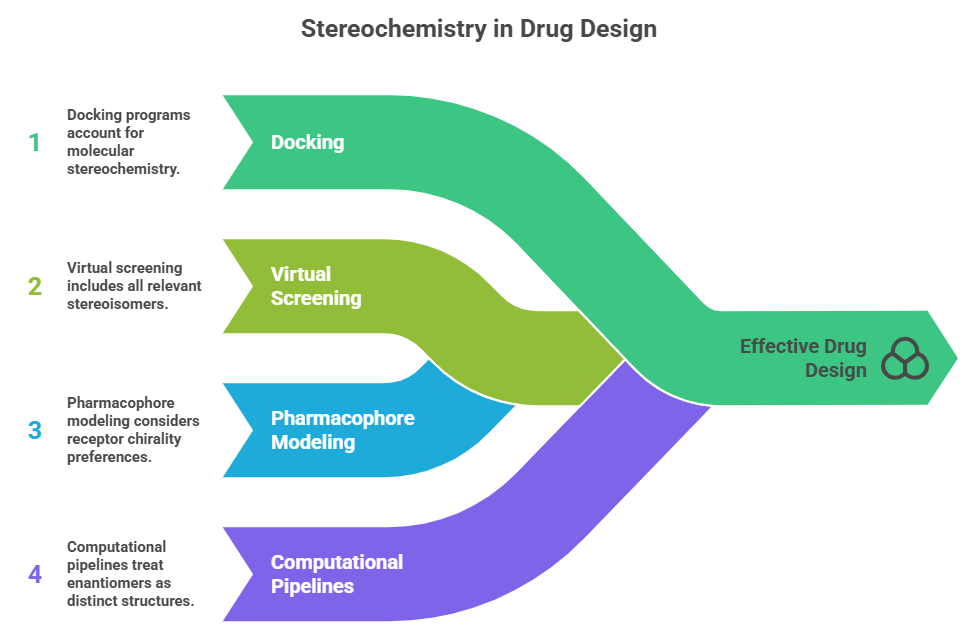
There’s also burgeoning research on using AI/ML to predict enantioselective synthesis outcomes (for catalysis) – tying to Part 10 content – but in design, AI could propose chiral molecules; the medicinal chemist must be aware of which enantiomer the AI suggests or to ensure training sets are properly enumerated with stereochemistry. The lifechemicals blog suggests that computational screening benefits from including chirality for potency differences.
Regulatory Guidelines (ICH/FDA/EMA)
From a development perspective, regulatory bodies require control of stereochemistry from the start. For example, ICH Q6A (spec for new drug substances) says if a drug is chiral, the enantiomeric purity (or diastereomer ratio) must be specified and tested. The FDA’s 1992 guidance (although a bit dated) is still a key reference: it recommends that sponsors: – Identify the stereochemical composition of the drug substance and product. – Develop chiral analytical methods (e.g., chiral HPLC) early to be able to monitor and quantify enantiomers in biological samples and in stability studies. – If developing a racemate, prove that both enantiomers’ pharmacokinetics and pharmacodynamics have been characterized and that you have a reason for using a mixture (like interconversion or both needed). Show that manufacture is consistent – e.g., no unintended epimerization or racemization occurring during process or shelf life (the Canada guidance excerpt indicates need to monitor enantiomeric purity during stability.
EMA has similar guidelines (they often refer to ICH guidelines). They also sometimes require separate toxicology studies on isolated enantiomers, especially if one enantiomer predominates in exposure or if a single is developed after a racemate (to ensure the removed enantiomer wasn’t mitigating some toxicity of the other – sometimes one enantiomer can competitively antagonize the other’s off-target effect, theoretically).
Chirality in Clinical Development
When a chiral drug enters trials, if it’s a racemate, often assays are run to see if both enantiomers are present in plasma and in what ratio (since metabolism might be enantioselective). If one enantiomer accumulates or has longer half-life, that might raise a flag to consider single enantiomer. For instance, verapamil is racemic but its enantiomers have different half-lives because of stereoselective metabolism. A developer might still market racemic verapamil, but they have to understand that S-verapamil (more potent at heart) is cleared faster, etc. Actually, with verapamil, that knowledge influences dosing but they didn’t switch to single enantiomer.
Case Study – Escitalopram vs Citalopram:
Citalopram is racemic; only S-enantiomer (escitalopram) is active as SSRI (R is ~30x weaker and might even counteract a bit). Lundbeck when developing escitalopram had to show in trials that 10 mg escitalopram is as effective as 20 mg citalopram, which they did. They could then claim improved tolerability and quicker onset (some subtle advantages). This influenced the decision to develop the single enantomer.
Another aspect – IP and market:
From a business standpoint, if a company holds patents on both racemic and single enantomer forms, they might plan a chiral switch. This influences R&D strategy: sometimes a single enant drug was in mind from the start but they’d initially sell racemate (like omeprazole was first sold racemic, later esomeprazole introduced). Some companies patent specifically the active enantomer early to block others from doing it later.
Summary (Part 9)
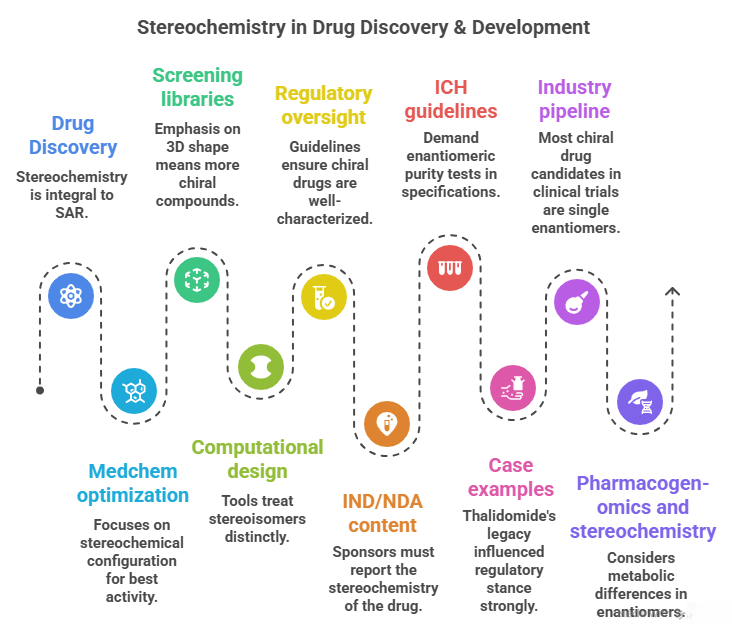
– Drug Discovery: Stereochemistry is integral to SAR. Hits are evaluated for stereochemistry – often only one enantiomer binds well. Medchem optimization focuses on the stereochemical configuration that yields best activity (the other config might be dropped unless there’s interest in dual activity). Eudismic ratios help quantify enantioselective SAR.
– Screening libraries: Emphasis on 3D shape means more chiral compounds. Strategies: either include both enantiomers as separate entries or screen racemates and deconvolute later. Each approach has cost/efficiency trade-offs. HTS equipment and analysis handle chirality by ensuring hits are confirmed with chiral resolution.
– Computational design: Tools treat stereoisomers distinctly. Virtual libraries are enumerated with stereocenters. Docking must consider stereochemistry to accurately predict binding. Pharmacophore models may encode chiral points (e.g., requiring a certain handed center for a H-bond orientation). AI approaches are also aware: for example, some generative chemistry algorithms can output molecules with or without specified stereochemistry; the user can enforce “only output chiral molecules” or “output racemic pairs as two entries”.
– Regulatory oversight: Strong guidelines ensure chiral drugs are well-characterized: known configuration, consistent production, and rational development strategy (choose single vs racemate based on data). The requirement for stereochemical analysis means drug developers implement chiral assays (HPLC, GC, capillary electrophoresis) early, often using them in preclinical and clinical sample analysis to see if any interconversion or disproportionation (one enantiomer disappearing relative to other) occurs.
– IND/NDA content: Sponsors must report the stereochemistry of the drug, how it’s controlled in manufacturing (e.g., if a chiral catalyst is used, how do they ensure consistent enantiomeric excess, or if they resolve, how they dispose of the other enantiomer, etc.), and data from any studies on individual enantiomer if relevant. The FDA might ask for toxicity data on the isolated enantiomer if the racemate is intended (especially if one enantiomer could be toxic as in some past cases).
– ICH guidelines: For example, ICH Q7 (manufacturing of APIs) and Q6A (specs) demand enantiomeric purity tests in specifications unless justified otherwise. ICH Eudralex also had a note: chirality considered a “critical quality attribute” for many drugs.
– Case examples: Thalidomide’s legacy influenced regulatory stance strongly: no assumptions that “it’s just the same” – each enantiomer should be treated as potentially different until proven otherwise. Warfarin’s development had to consider stereochemistry in dosing (the S vs R potency difference is reflected in dosing differences between patients with varying metabolism). In discovery of modern anticoagulants (like dabigatran), they chose single stereochemistry from the start.
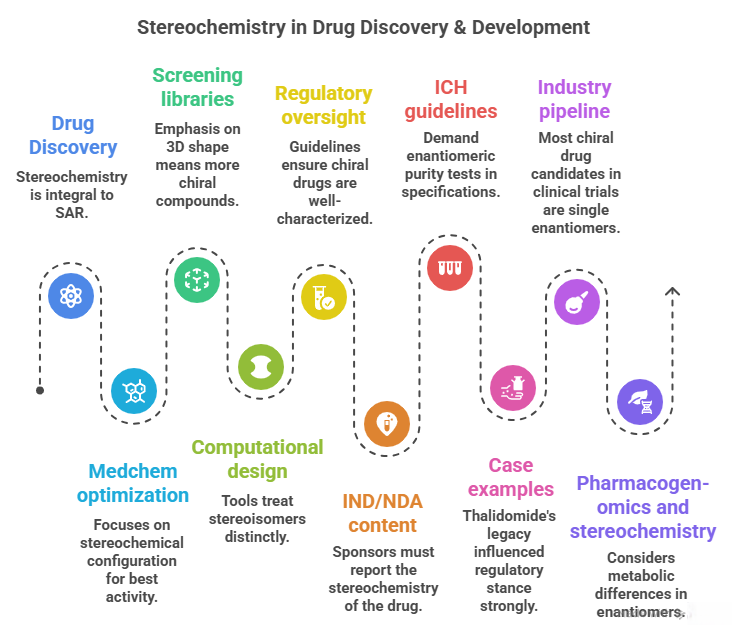
– Industry pipeline: Today, most chiral drug candidates in clinical trials are single enantiomers (unless there’s a specific reason to have both). That means medicinal chemists either synthesize them asymmetrically or resolve them during preclinical stage. It’s part of the decision gate: if you have a racemic lead, can you easily get one pure enantiomer? If not, is the project viable? They might invest in developing a route or using contract chemists to do resolution for small scale.
– Pharmacogenomics and stereochemistry: Another nuance – as seen with omeprazole, polymorphisms in metabolism made one enantiomer’s clearance more variable. Similarly, some people metabolize one enantiomer differently (CYP2D6 metabolizes one propranolol enantiomer faster). This can lead to differences in drug response or interactions, which regulators consider – sometimes recommending dosing adjustments if one enantiomer in racemate builds up in certain genotypes.
Suggested Reading
Wenxiu Wei et al., (0220), “Fsp3: A new parameter for drug-likeness” Drug Discovery Today Volume 25, Issue 10, October 2020, Pages 1839-1845 https://doi.org/10.1016/j.drudis.2020.07.01
Mei, H. et al. (2016). “Drug-like properties: Guidelines for maintaining stereochemical diversity in screening libraries.” J. Med. Chem., 59(13), 6359–6363. (Importance of stereochemical diversity and suggestions to med chem practitioners.)
https://lifechemicals.com/screening-libraries/fsp3-enriched-library
Keserü, G. M., & Makara, G. M. (2006). “The influence of lead discovery strategies on the properties of drug candidates.” Nat. Rev. Drug Discov., 5, 203–212. (Contains discussion on 3D character of leads and how discovery approaches yield different stereochemical outcomes.)
ICH Q6A: “Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances.” (Section on chiral substances outlines expectations for control of enantiomeric purity.)
FDA Guidance for Industry: “Stereochemical Issues in Drug Development” (1992). Although older, it comprehensively discusses how to plan studies for chiral drugs – still a useful reference point. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/development-new-stereoisomeric-drugs
Chirality in Drug Research (Topics in Current Chemistry vol. 243) – contains chapters on stereochemistry in screening and drug design, plus regulatory considerations. One chapter by FDA authors might detail how they review stereochemical data in NDAs.
Silvestri IP, Colbon PJJ. The Growing Importance of Chirality in 3D Chemical Space Exploration and Modern Drug Discovery Approaches for Hit-ID: Topical Innovations. ACS Med Chem Lett. 2021 Jul 16;12(8):1220-1229. doi: 10.1021/acsmedchemlett.1c00251.
Ceramella, J.; Iacopetta, D.; Franchini, A.; De Luca, M.; Saturnino, C.; Andreu, I.; Sinicropi, M.S.; Catalano, A. A Look at the Importance of Chirality in Drug Activity: Some Significative Examples. Appl. Sci. 2022, 12, 10909. https://doi.org/10.3390/app122110909
Eric Francotte, W. Lindner. Weinheim, Chirality in drug research. 2006, Wiley-VCH. ISBN 978-3-527-60943-7. OCLC 163578005
