“Before the world looked closely at molecular mirror images, the pharmaceutical landscape was filled with blind spots -this is the story of how chirality found its voice.”
Introduction
The phenomenon of chirality, where molecules exist as non-superimposable mirror images, permeates the natural world. In biological systems, chirality is fundamental: proteins, enzymes, and receptors are inherently stereospecific, responding differently to the two mirror-image forms, or enantiomers, of a compound. In pharmacology, these subtle molecular differences can translate into significant divergences in therapeutic efficacy, safety, and metabolism (Caldwell, 1995; Waldeck, 2003). Yet, for much of pharmaceutical history, this vital detail was largely overlooked.
Prior to the 1990s, the pharmaceutical industry and regulatory agencies treated most chiral drugs as racemic mixtures -equimolar blends of two enantiomers – without demanding rigorous characterization of the individual components. It was assumed, often incorrectly, that the effects of a racemate simply represented the sum of its parts. As drug discovery became more sophisticated and tragic events like the thalidomide crisis revealed the devastating consequences of neglecting stereochemistry, the tide began to shift.
This blog explores the pre-1990s regulatory landscape for chiral drugs, the early scientific clues about enantiomeric differences, and the slow awakening that set the stage for the seismic regulatory transformations of the 1990s.
Understanding Chirality: The Basics
Chirality, from the Greek word for hand (χειρ, kheir), describes objects that are mirror images but not superimposable – like left and right hands. Molecules with this property possess one or more chiral centers, typically a carbon atom bonded to four different substituents, resulting in two distinct enantiomers. In pharmacology, these mirror-image molecules may interact differently with the body’s chiral environments. Often, one enantiomer (the eutomer) is primarily responsible for the desired therapeutic effect, while the other (distomer) may be inactive, less active, or even produce adverse effects (Jamali & Brocks, 1990). However, until relatively recently, the clinical implications of this molecular duality were not fully appreciated.
Early Practices: Racemic Drugs as the Norm
Throughout the 20th century, technical limitations made it difficult to isolate and produce pure enantiomers on a commercial scale. As a result, most chiral drugs were developed and marketed as racemic mixtures. There were practical reasons for this approach: enantiomers have identical physical properties in achiral environments (melting point, solubility, etc.), making separation complex and expensive (Wang, Sun, & Hao, 2005).
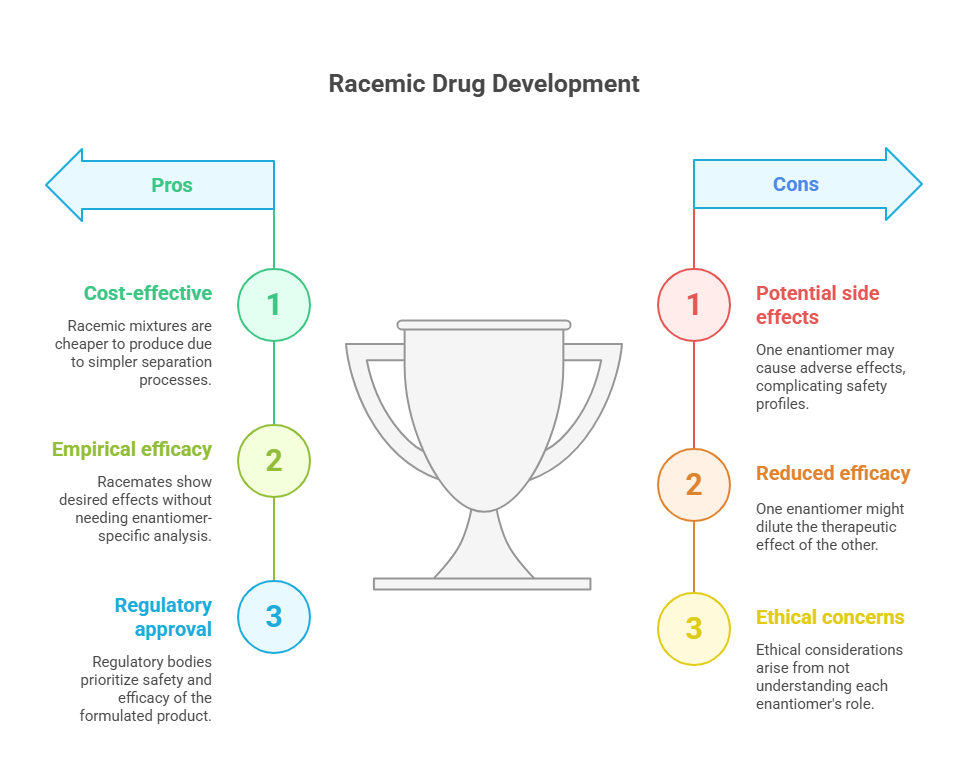
For drug developers, the guiding principle was empirical efficacy. If a racemate showed the desired therapeutic effect and acceptable safety in clinical trials, there was little incentive to dissect the contribution of each enantiomer. Regulatory bodies, similarly, did not require such analyses. Approval processes focused on the safety and efficacy of the formulated product, with minimal attention to underlying stereochemistry.
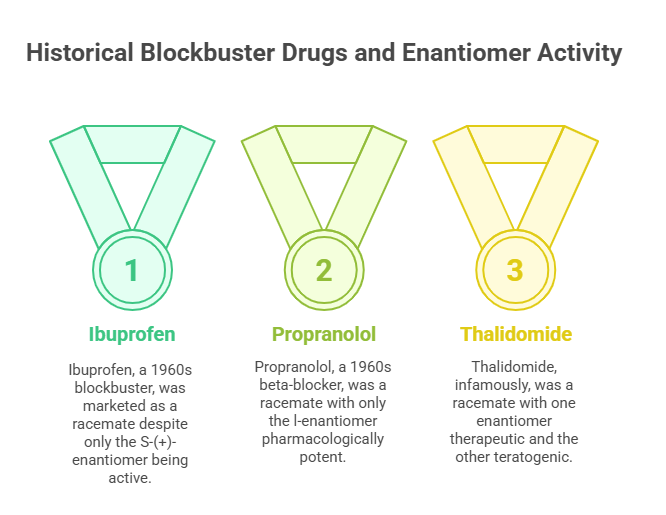
Several blockbuster drugs exemplify this historical pattern:
- Ibuprofen, introduced in the 1960s, was marketed as a racemate even though only the S-(+)-enantiomer is an active cyclooxygenase inhibitor (Jamali & Brocks, 1990).
- Propranolol, a beta-blocker approved in the 1960s, was a racemate despite only the l-enantiomer being pharmacologically potent at beta-adrenergic receptors (Waldeck, 2003).
- Thalidomide, infamously, was developed and administered as a racemate without recognizing that one enantiomer was therapeutic while the other was teratogenic (Agranat, Caner, & Caldwell, 2002).
In these and numerous other cases, the racemate was treated as a single entity, with no regulatory insistence on enantiomeric characterization.
The Scientific Clues: When Enantiomers Behaved Differently
Despite widespread reliance on racemates, the scientific literature offered growing hints throughout the mid-20th century that enantiomers could behave differently:
- In thalidomide, administered to pregnant women for morning sickness in the late 1950s, the R-enantiomer provided sedative effects, while the S-enantiomer was responsible for severe birth defects (McBride, 1961). Although thalidomide racemizes in vivo (making single-enantiomer formulations ineffective for avoiding teratogenicity), the tragedy dramatically underscored the biological significance of stereochemistry.
- Penicillamine, an anti-arthritic drug, exhibits dramatically different toxicity profiles between its D- and L-forms, with the D-isomer associated with greater adverse effects (Wang et al., 2005).
- Studies on propranolol showed that while the S-enantiomer was a potent beta-adrenergic blocker, the R-enantiomer was much less effective (Waldeck, 2003).
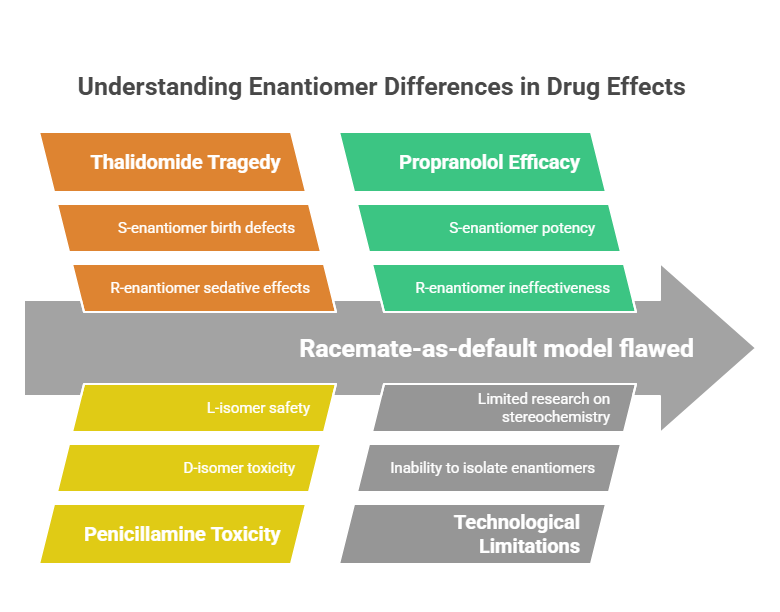
These findings were piecemeal and, initially, lacked the technological means to influence mainstream drug development. Nevertheless, a body of evidence was quietly accumulating that suggested the racemate-as-default model was scientifically flawed.
The Technological Barrier: Why Pure Enantiomers Were Rare
Even as scientists recognized enantiomer-specific activity, synthesizing or isolating single enantiomers at scale was a formidable challenge. Traditional methods like resolution with chiral auxiliaries or repeated recrystallization were inefficient and costly (Agranat et al., 2002).
Moreover, regulatory frameworks of the time did not incentivize companies to invest in enantioselective synthesis. Without a regulatory mandate, and given the commercial viability of racemic products, the pharmaceutical industry largely continued developing racemates.
The Regulatory Blind Spot: No Guidance on Chirality
Before the early 1990s, regulatory authorities like the U.S. Food and Drug Administration (FDA), European national agencies, and Japan’s Ministry of Health and Welfare did not have formal guidance addressing the development of stereoisomeric drugs. Applications for new drugs rarely discussed enantiomeric composition unless it had a direct impact on efficacy or toxicity.
The general regulatory view was that the racemic product’s overall clinical profile was sufficient to determine safety and efficacy. As long as the clinical trials demonstrated that the racemic mixture worked and was safe, there was no requirement to investigate whether one enantiomer was superior – or harmful.
This gap persisted despite increasing academic awareness of stereochemical issues. It reflected the practical realities of the time: separating enantiomers was difficult, analytical methods to characterize them were limited, and regulatory science had not yet caught up with academic advances.
The Wake-Up Calls: Why Change Became Necessary
For years, the intricate world of stereochemistry remained overlooked by scientists in drug development, largely because there simply wasn’t the technology to examine its three-dimensional impacts. But two pivotal events brought these issues into sharp focus. The first was the tragic thalidomide disaster, which underscored the devastating consequences of neglecting stereochemical nuances. The second was the release of a groundbreaking manuscript in 1984, titled “Stereochemistry, a basis of sophisticated nonsense in pharmacokinetics and clinical pharmacology“ (Ariens, 1984). This provocative piece, along with a series of follow-up articles,(Ariens, 1986; 1987a,b,c; 1989), called out the prevailing practice of studying racemic drugs as a whole – ignoring how each enantiomer uniquely contributes to the drug’s behavior. These publications sparked a wave of debate and reflection, stirring fresh conversations in academic, industrial, and regulatory circles about the critical role stereochemistry plays in drug research and development.
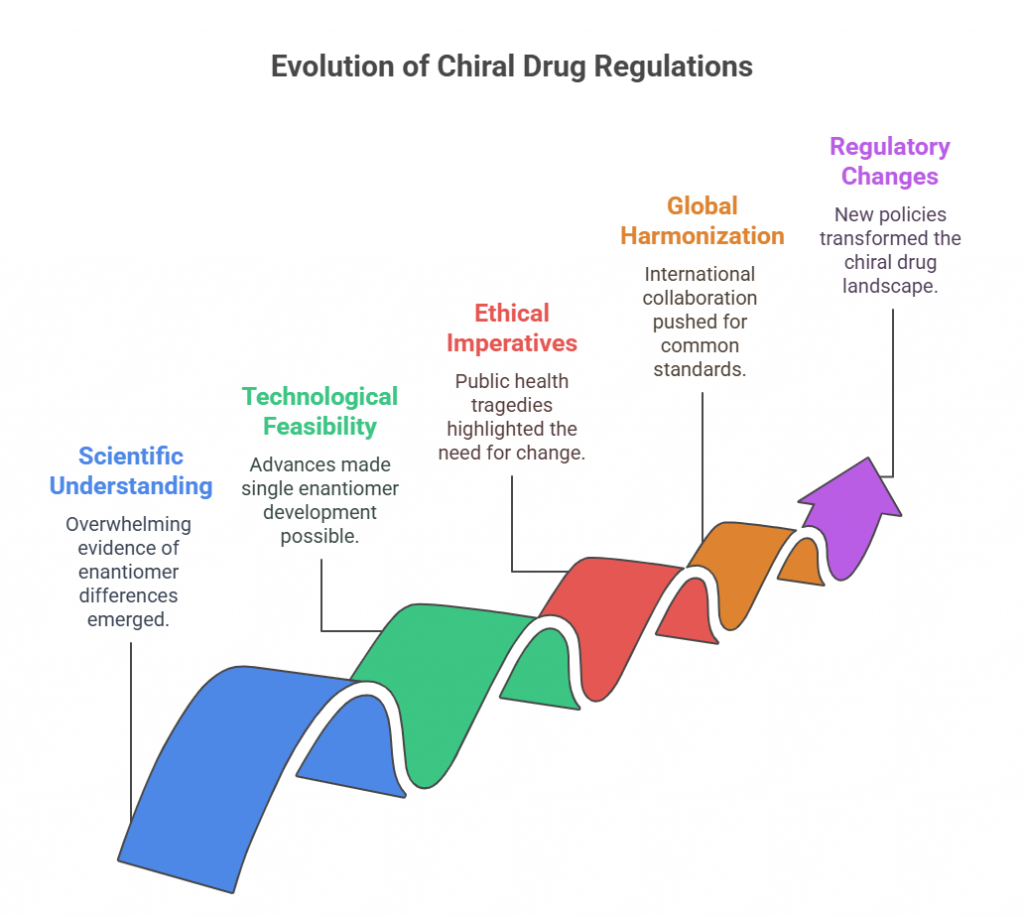
By the late 1980s, several converging factors made it clear that a paradigm shift was needed:
- Scientific understanding: There was overwhelming evidence that enantiomers could have distinct pharmacological and toxicological profiles.
- Technological feasibility: Advances in asymmetric synthesis, chiral chromatography, and stereoselective bioanalysis made it possible to develop and characterize single enantiomers at commercial scale (Hancu & Modroiu, 2022).
- Ethical imperatives: Public health tragedies like thalidomide reminded regulators of the stakes involved in stereochemical ignorance.
- Global harmonization pressures: As international collaboration in regulatory science grew (eventually leading to the International Council for Harmonisation, or ICH), there was increasing impetus to establish common standards, including in stereochemistry.
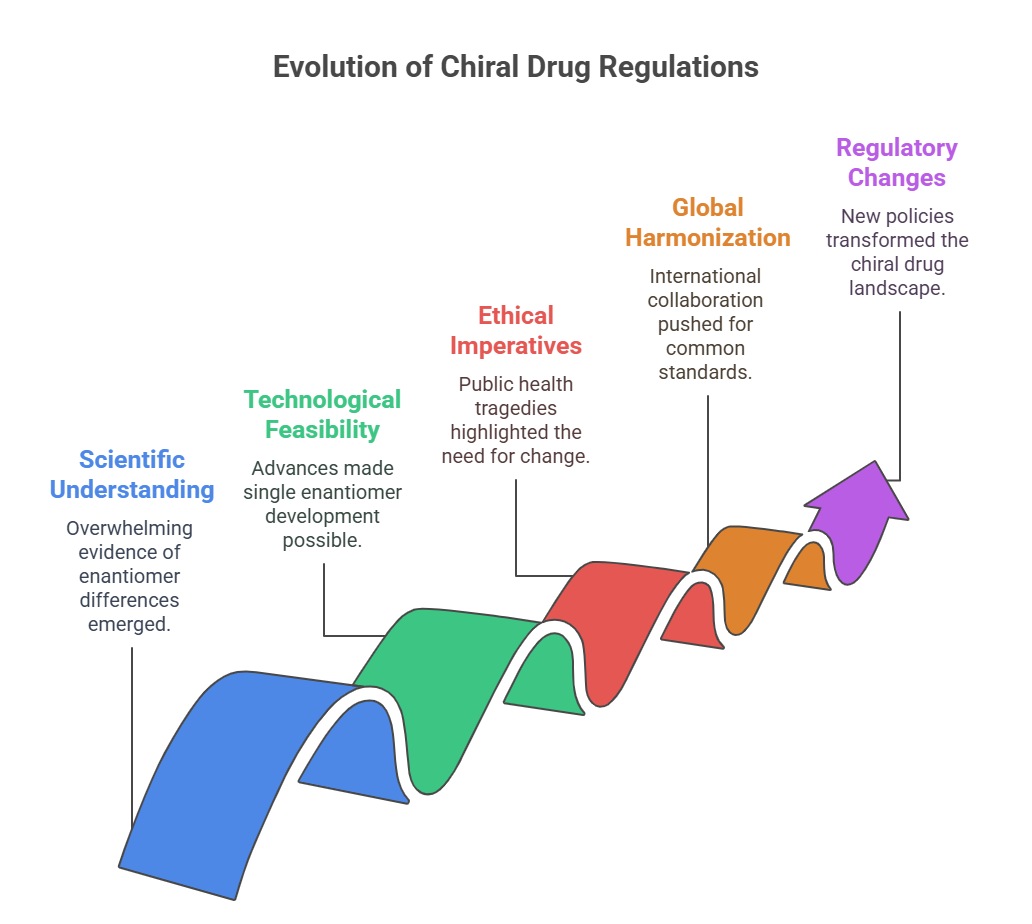
Against this backdrop, regulatory agencies began to reconsider their approach to chiral drugs. The academic debate spilled into policy discussions, culminating in pivotal regulatory changes that would forever alter the landscape.
Conclusion
The history of chiral drugs before the 1990s is a study in scientific humility. It reveals how technological limitations, regulatory inertia, and commercial pragmatism can overshadow important biological truths. Yet, it also shows how scientific knowledge eventually forces change, even in the most entrenched systems.
As we shall see in the next episode, the 1990s heralded a revolution. The FDA, EMA, and other agencies would soon issue groundbreaking policies that demanded enantiomeric characterization, justified the development of single-enantiomer drugs, and reshaped the trajectory of pharmaceutical science.
The mirror images are no longer invisible.
What is in the next episode?
“What happens when regulators catch up with science? Join us for Episode 2: The Great Shift: How the 1990s Redefined Stereochemical Standards, where we dive into the transformative policies that forever changed chiral drug development.“
References
Ariens, E. J. (1984). “Stereochemistry, a basis for sophisticated nonsense in pharmacokinetics and clinical pharmacology”. European Journal of Clinical Pharmacology. 26 (6): 663–668. doi:10.1007/bf00541922.
Ariëns, E.J. (1986). “Chirality in bioactive agents and its pitfalls”. Trends in Pharmacological Sciences. 7: 200–205. doi:10.1016/0165-6147(86)90313-5.
Ariëns, Everardus J. (1987a). “Stereochemistry in the analysis of drug-action. Part II”. Medicinal Research Reviews. 7 (3): 367–387. doi:10.1002/med.2610070305.
Ariëns, E.J (1987b). “Implications of the Neglect of Stereochemistry in Pharmacokinetics and Clinical Pharmacology”. Br. J. Clin. Pharmacol. Ther. 21 (10): 827–829. doi:10.1177/106002808702101013.
- Ariëns, Everd J. (1987c). “Implications of the Neglect of Stereochemistry in Pharmacokinetics and Clinical Pharmacology”. Drug Intelligence & Clinical Pharmacy. 21 (10): 827–829. doi:10.1177/106002808702101013.
Ariëns, E. J. (1989). “Stereoselectivity in the Action of Drugs”. Pharmacology & Toxicology. 64 (4): 319–320. doi:10.1111/j.1600-0773.1989.tb00655.x. .
Agranat, I., Caner, H., & Caldwell, J. (2002). Putting chirality to work: The strategy of chiral switches. Nature Reviews Drug Discovery, 1(10), 753–768.
Caldwell, John (1995). “Chiral Pharmacology and the regulation of new drugs”. Chemistry and Industry. 6: 176–179.
Hancu, G., & Modroiu, A. (2022). Chiral switch: Between therapeutical benefit and marketing strategy. Pharmaceuticals, 15(2), 240.
Jamali, F., & Brocks, D. R. (1990). Clinical pharmacokinetics of ketoprofen and its enantiomers. Clinical Pharmacokinetics, 19(3), 197–217.
McBride, W. G. (1961). Thalidomide and congenital abnormalities. The Lancet, 278(7216), 1358.
Waldeck, B. (2003). Three-dimensional pharmacology: Effects of chirality on pharmacokinetics and pharmacodynamics. Pharmacology & Therapeutics, 95(1), 1–12.
Wang, G., Sun, J., & Hao, H. (2005). Enantioselective pharmacokinetics of ibuprofen and involved mechanisms. Drug Metabolism Reviews, 37(1), 215–234.
Further Reading
https://en.wikipedia.org/wiki/Chiral_analysis
