Synopsis
The thalidomide tragedy is one of the most infamous episodes in pharmaceutical history—yet its stereochemical secrets are still being unraveled. A fascinating article by Tokunaga E, Yamamoto T, Ito E, and Shibata N., published in Scientific Reports (Nature Research, 2018; https://www.nature.com/articles/s41598-018-35457-6), sheds new light on the phenomenon through the lens of the self-disproportionation of enantiomers, the physical chemistry of chirality.
Thalidomide’s tragic past is linked to its two “mirror-image” forms. One form, the S-enantiomer, can cause severe birth defects, while the other, the R-enantiomer, appears harmless. But here’s the puzzle: inside the body, the two forms can switch into each other—so giving R-thalidomide should still make some harmful S-thalidomide. Yet animal tests with R-thalidomide didn’t cause defects. This study shows why. In water-like conditions, mixtures of R and S “self-sort” — the harmless form stays dissolved, while equal mixtures of R and S clump together into crystals and drop out of solution. Because these racemic clumps are poorly soluble, they likely can’t reach target tissues. This means that even if some S-thalidomide forms in the body, much of it gets locked away before it can do harm.
But let’s face it—in today’s fast-paced, digital-first world, even the most dedicated medicinal and chiral chemists may not have time to read an entire research paper cover to cover. That’s why this blog takes the deep science and dissects, decodes, and demystifies it—transforming the complex into a visually engaging, easy-to-grasp story. The goal? To make learning not only faster but also more impactful and memorable.
The Thalidomide Paradox: How a Chiral Twist May Explain a 60-Year Mystery
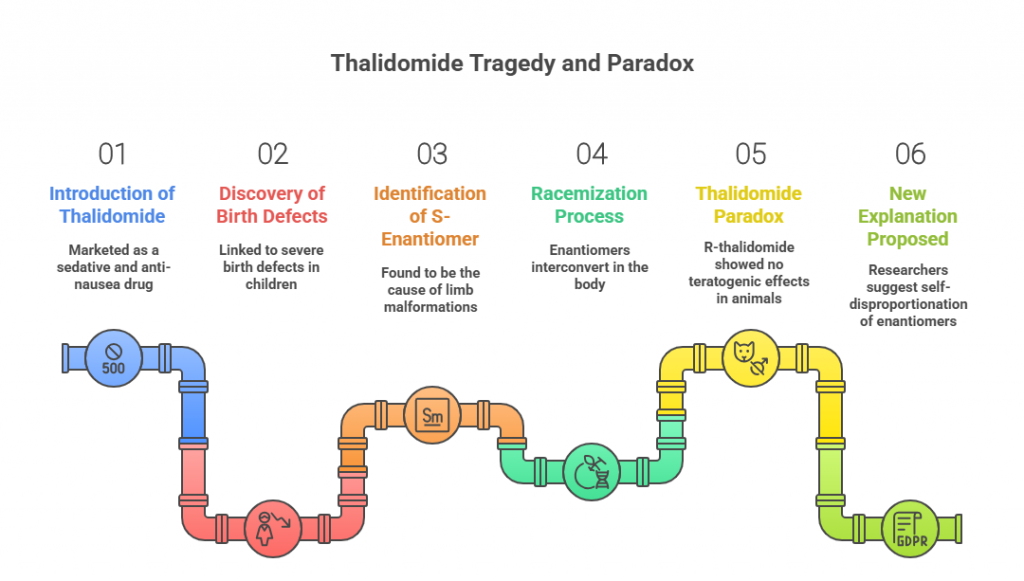
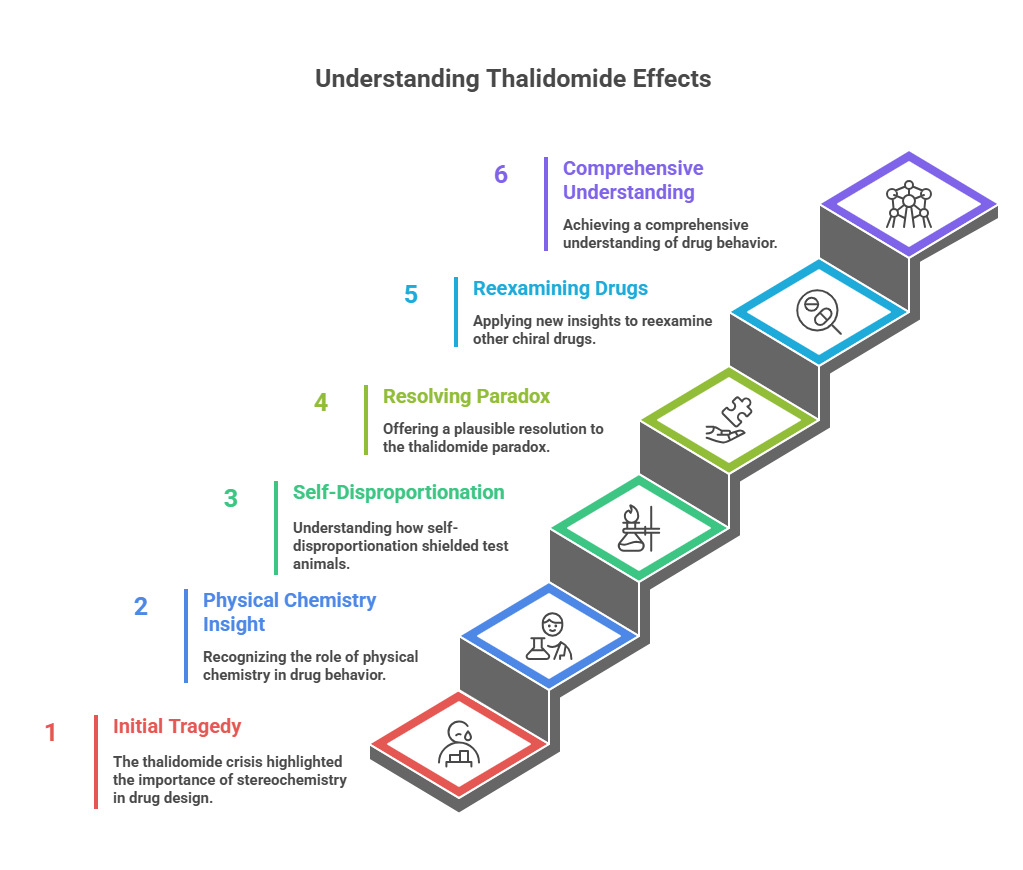
Few drugs have etched themselves into medical history as starkly as thalidomide. Introduced in the late 1950s as a sedative and anti-nausea pill — even recommended to pregnant women — it seemed a harmless innovation. But it soon became the center of one of the greatest pharmaceutical tragedies of the 20th century, linked to thousands of cases of severe birth defects.
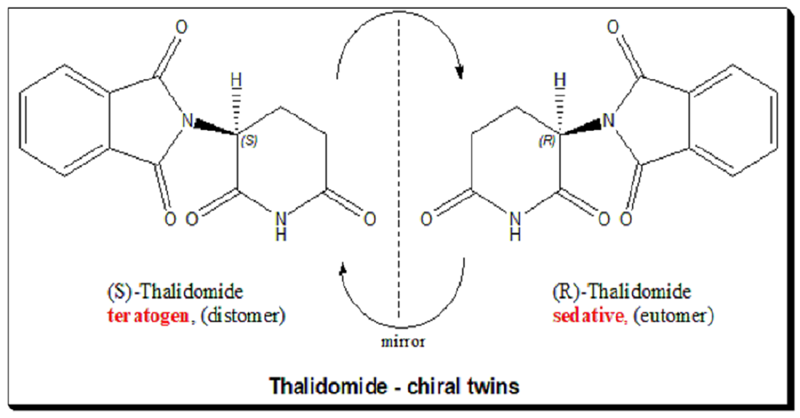
The story has been retold countless times: the culprit was the drug’s S-enantiomer, a molecular “hand” that caused limb malformations, while its mirror image — the R-enantiomer — was not teratogenic. It seemed like a lesson in chirality: if only the R-form had been marketed, the disaster might have been avoided.
But then came the problem. Both enantiomers interconvert in the body — a process called racemization. That means that even if you give pure R-thalidomide, some of it turns into the harmful S-form in vivo. Yet, oddly, animal experiments using R-thalidomide showed no teratogenic effects. This long-standing contradiction became known as the “thalidomide paradox.”
Now, researchers Etsuko Tokunaga, Takeshi Yamamoto, Emi Ito, and Norio Shibata propose a compelling new explanation — one that hinges on an underappreciated process called self-disproportionation of enantiomers (SDE). Their findings don’t just help solve a decades-old puzzle; they also raise caution flags for many other chiral drugs.
From Mirror Images to Molecular Fate
Thalidomide is a chiral molecule with a single stereogenic carbon atom, giving rise to two non-superimposable mirror images — the R and S enantiomers. In the racemic drug (a 50:50 mixture), the two enantiomers interact differently with biological targets. In 1979, Blaschke and colleagues discovered that only S-thalidomide caused birth defects in animals. Read more @ <https://chiralpedia.com/blog/thalidomide-tragedy-the-story-and-lessons/> and <https://chiralpedia.com/blog/thalidomide/>
The easy conclusion was: use R-thalidomide instead. But there was a catch. R and S readily interconvert under physiological conditions, with half-lives of just hours in buffer and blood. That meant giving R-thalidomide should inevitably produce S-thalidomide in vivo — yet, paradoxically, teratogenicity still wasn’t observed in the “pure” R dosing experiments.
For decades, this contradiction remained unresolved. Then, a key clue emerged from studies of thalidomide’s binding target: the protein cereblon (CRBN). In 2010, Handa’s group identified CRBN as the molecular docking site linked to teratogenicity. Later, structural work confirmed that both enantiomers can bind to CRBN, but the S-form does so much more tightly — with about ten times higher affinity — and adopts a more “natural” ring conformation in the binding pocket. This solidified that only S-thalidomide is teratogenic.
Still, the question remained: if R turns into S inside the body, why don’t we see teratogenicity when R is administered?
A Physical Chemistry Twist: Self-Disproportionation of Enantiomers
The answer, Tokunaga and colleagues suggest, lies in a process more familiar to physical chemists than to pharmacologists: self-disproportionation of enantiomers (SDE).
Coined in 2006, SDE describes the spontaneous separation of an enantiomeric mixture into fractions with different enantiomeric purities. In practical terms, if you have a partially enriched chiral mixture in solution, it can “self-sort” — the purer enantiomer stays dissolved, while a near-racemic portion crystallizes or precipitates out.
The authors noticed striking differences in the solubility of thalidomide’s pure enantiomers versus the racemate:
- (R)- and (S)-thalidomide are both about 5.5 times more soluble in water than the racemic form.
- X-ray crystallography showed why: the racemate forms a tightly packed heterodimer linked by two hydrogen bonds, making it less soluble. The enantiomers, in contrast, can form more soluble monomers or homodimers.
This meant that in a biological setting, partially racemized thalidomide might behave like this:
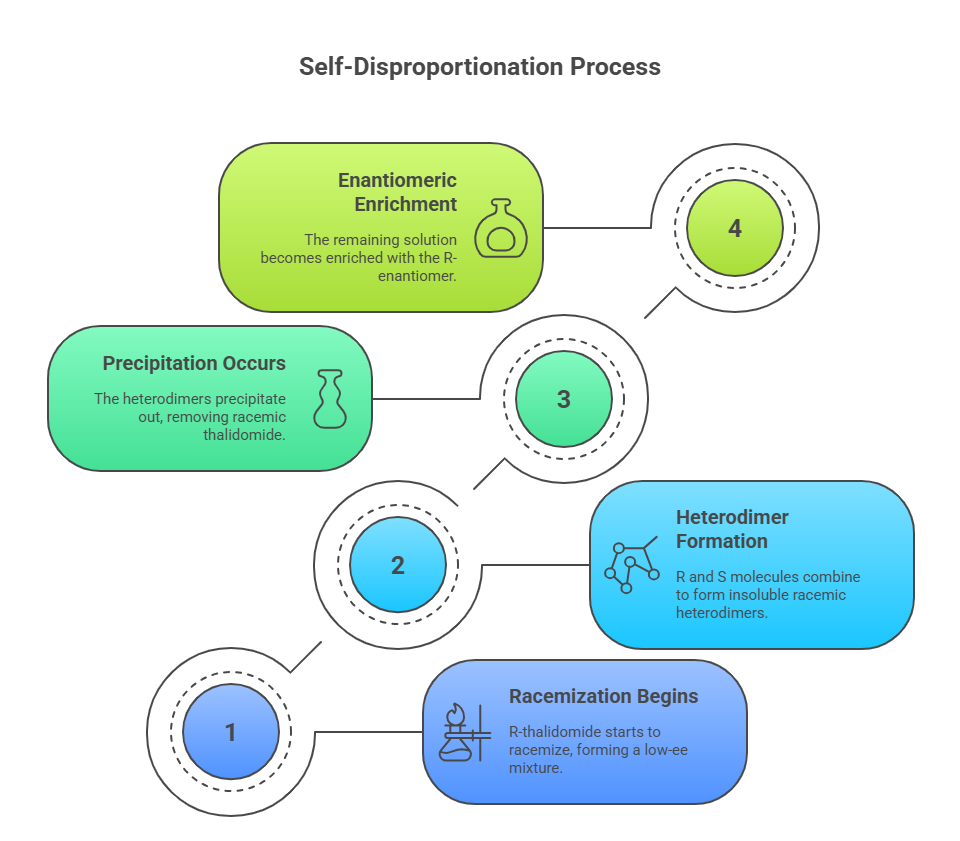
- R-thalidomide in vivo begins to racemize, producing a low-ee mixture (say, 20% enantiomeric excess).
- The R and S molecules form highly insoluble racemic heterodimers.
- These precipitate out — effectively removing racemic thalidomide from the biological fluid.
- The remaining dissolved fraction becomes enantiomerically enriched again — in this case, still rich in R.
If the harmful S-form preferentially drops out with the racemate, it never accumulates in solution to levels high enough to cause teratogenic effects — even though racemization is happening.
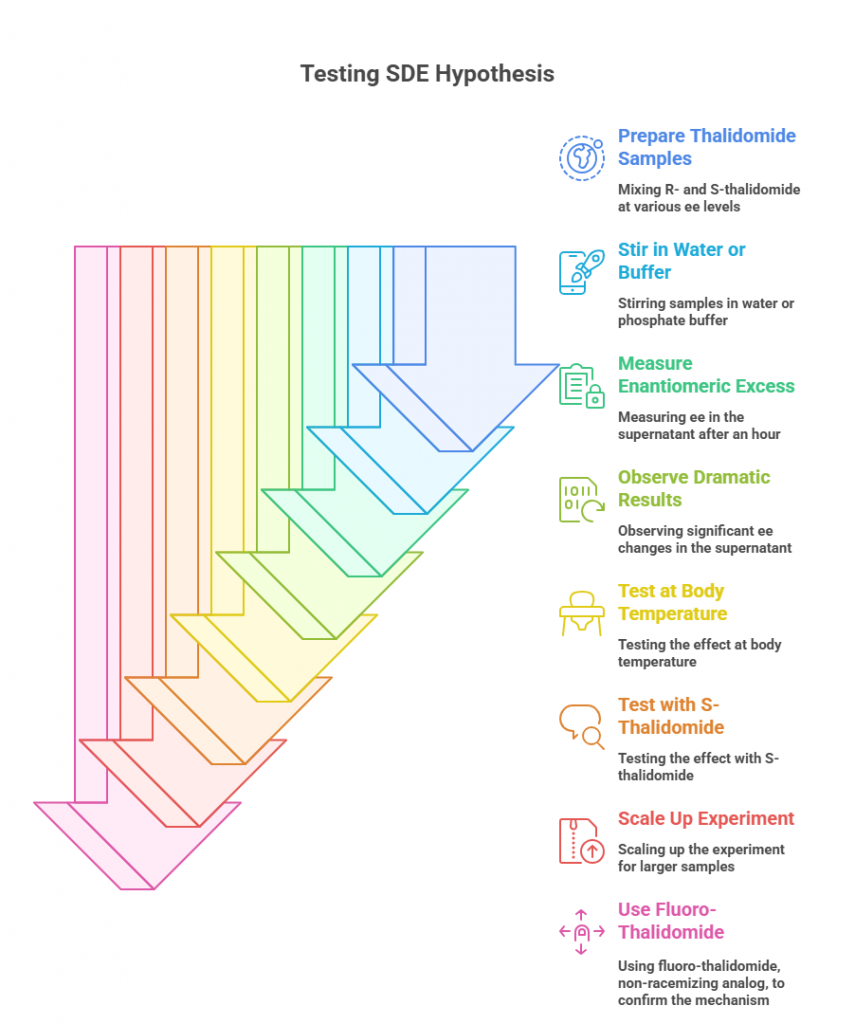
Testing the Hypothesis
The team tested SDE under a range of lab conditions designed to mimic physiological environments. They mixed R- and S-thalidomide at various starting enantiomeric excesses, stirred them in water or phosphate buffer, and measured the enantiomeric excess in the supernatant after an hour. The results were dramatic:
- In water at room temperature, a 20% ee sample of R-thalidomide yielded a 97% ee supernatant after one hour, with a large amount of low-ee precipitate.
- The effect was robust at body temperature (37°C) and in phosphate buffer at physiological pH.
- Similar behavior occurred with S-thalidomide and at different starting ee values.
- Scaling up the experiment produced consistent results: the soluble fraction became highly enriched, while the precipitate was nearly racemic.
They even repeated the experiment with fluoro-thalidomide, a non-racemizing analog, and found the same precipitation-driven enrichment — showing that the process is driven by solubility and molecular packing, not just racemization kinetics.
Reframing the Thalidomide Paradox
With this in mind, the paradox resolves itself:
- Yes, R-thalidomide racemizes in vivo.
- But the resulting racemic fraction precipitates out as insoluble heterodimers and is essentially removed from circulation.
- What remains in solution — and thus able to interact with biological targets — is predominantly the originally administered enantiomer.
In the animal experiments that led Blaschke to conclude R-thalidomide was non-teratogenic, the doses were large enough that precipitation would be likely. The oral absorption of racemic thalidomide is also known to be slower than that of the pure enantiomers, consistent with precipitation hindering uptake.
Implications Beyond Thalidomide
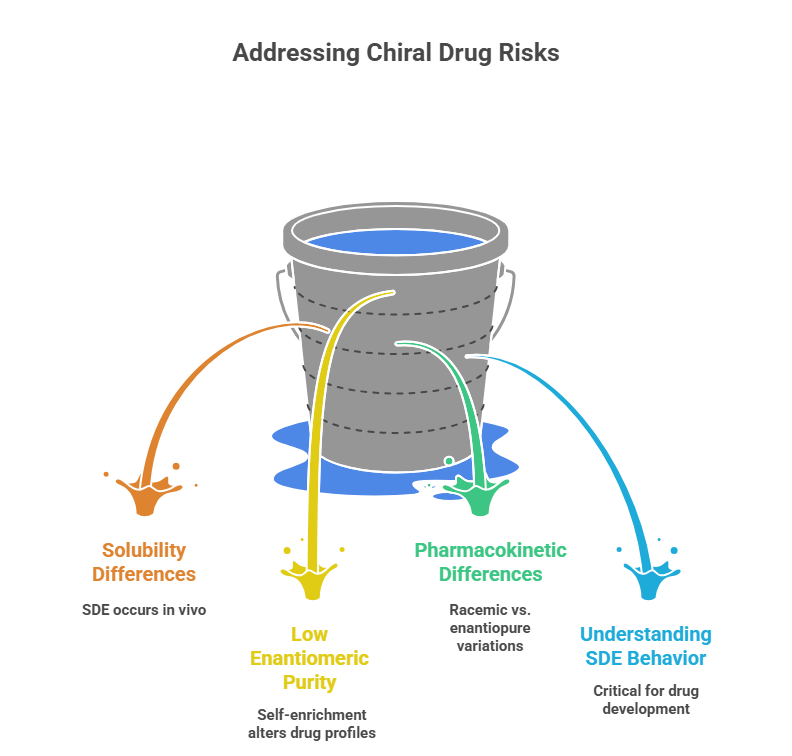
This insight isn’t just a historical curiosity. It has broader implications for any chiral drug:
- SDE can occur in vivo if the enantiomers differ in solubility from the racemate.
- Drugs with low enantiomeric purity might self-enrich in the body, altering both efficacy and toxicity profiles.
- This could lead to unexpected differences in pharmacokinetics between racemic and enantiopure formulations.
- In drug development, especially for chiral drugs with potential toxicity in one enantiomer, understanding SDE behavior may be critical.
The authors caution: “Chiral drugs with low enantiomeric purity should be used only with extreme care due to potential SDE processes in biological systems.”
From Tragedy to Molecular Insight
The thalidomide story began as a warning about the dangers of underestimating stereochemistry in drug design. This new chapter shows that the physical chemistry of chirality — not just the biology — can profoundly shape drug behavior in the body.
By revealing how self-disproportionation may have shielded test animals from thalidomide’s teratogenic effects in certain experiments, Tokunaga and colleagues not only offer the most plausible resolution yet to the “thalidomide paradox” but also open the door to reexamining other chiral drugs through the same lens.
In the end, the lesson is clear: the handedness of molecules is only part of the story — how those hands join, fold, and fall out of solution can be just as decisive in shaping a drug’s fate.
References:
Tokunaga, E., Yamamoto, T., Ito, E. et al. Understanding the Thalidomide Chirality in Biological Processes by the Self-disproportionation of Enantiomers. Sci Rep 8, 17131 (2018). https://doi.org/10.1038/s41598-018-35457-6 and references therein. https://www.nature.com/articles/s41598-018-35457-6
