“Science may be universal, but regulation wears many faces. Let’s journey through the world of chiral drug approval across continents.”
Introduction
The 1990s witnessed a revolutionary shift in the regulatory treatment of chiral drugs, spurred by major agencies like the U.S. FDA and European Medicines Agency (EMA). However, the global regulatory landscape was -and still is -far from homogeneous. While scientific consensus around the importance of stereochemistry has largely been achieved, regulatory expectations, procedural details, and enforcement rigor vary across jurisdictions.
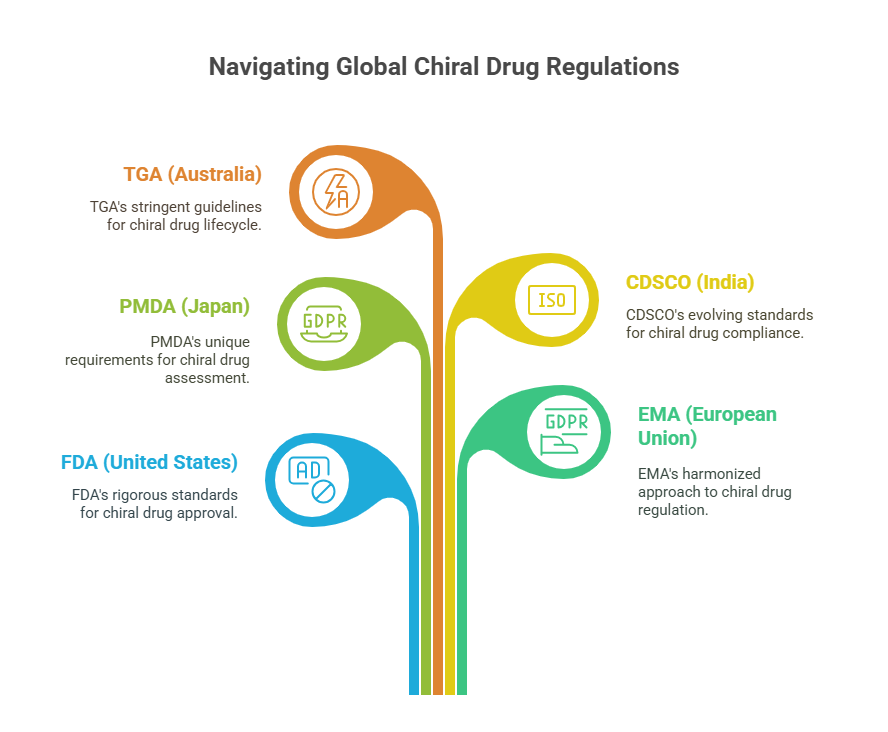
In this episode, we critically examine how major health authorities -including the FDA (United States), EMA (European Union), PMDA (Japan), CDSCO (India), and TGA (Australia) -approach the evaluation, approval, and lifecycle management of chiral drugs. Despite convergences encouraged by international harmonization efforts like ICH, key differences remain in how enantiomeric purity, bioequivalence, toxicology, and manufacturing controls are assessed.
Understanding these differences is crucial for global pharmaceutical strategy, regulatory compliance, and ultimately, patient safety.
The Common Foundation: Global Harmonization Efforts
The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) has played a pivotal role in standardizing the expectations around chiral drugs, especially through:
- ICH Q6A: Specifications for impurities, including enantiomeric impurities (ICH, 1999).
- ICH E5: Ethnic sensitivity and pharmacokinetic evaluation across populations (ICH, 1998).
- ICH M4: Common Technical Document (CTD) format requiring stereochemical data in Module 3 (Quality) and Module 5 (Clinical Study Reports).
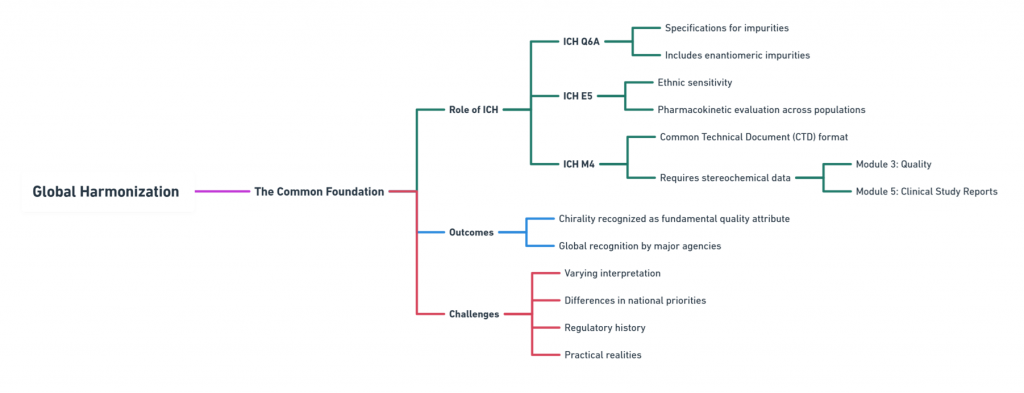
Thanks to ICH, all major agencies today recognize chirality as a fundamental quality attribute. However, interpretation and implementation vary based on national priorities, regulatory history, and practical realities.
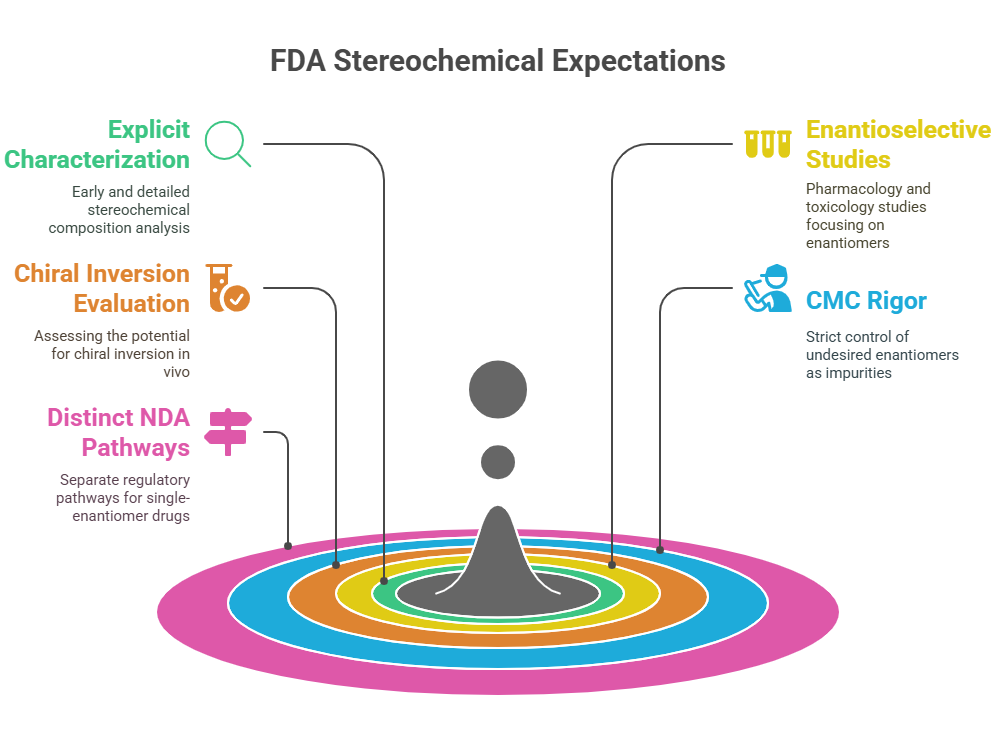
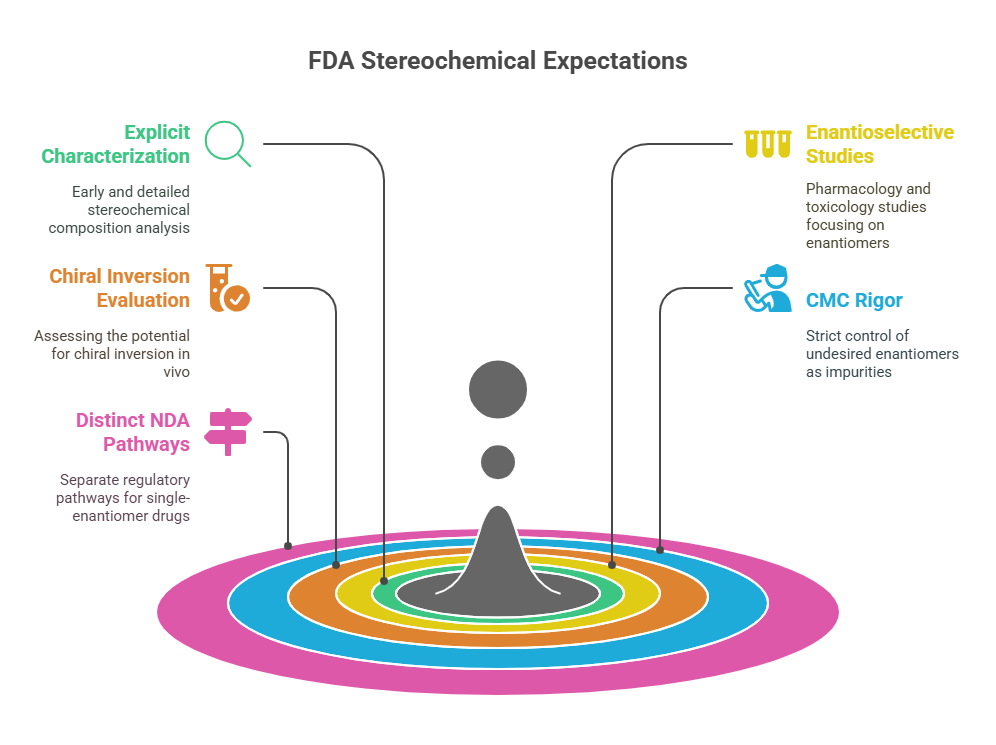
United States: FDA’s Rigorous Stereochemical Expectations
The U.S. FDA set the global standard early with its 1992 Policy Statement for the Development of New Stereoisomeric Drugs (FDA, 1992).
Key FDA Expectations
- Explicit characterization of stereochemical composition early in development.
- Enantioselective pharmacology and toxicology studies if enantiomers differ significantly.
- Evaluation of chiral inversion potential in vivo.
- CMC (Chemistry, Manufacturing, and Controls) rigor: controlling the undesired enantiomer as an impurity.
- Distinct NDA pathways for single-enantiomer drugs derived from racemates (“chiral switches”) (FDA, 1992).
Practical Observations
- The FDA expects strong scientific justification if a company chooses to market a racemate rather than a purified enantiomer.
- For chiral switches, superiority claims must be backed by robust clinical data (e.g., esomeprazole vs. omeprazole).
Strengths: Clear guidance, enforcement consistency, high scientific expectations.
Challenges: High data burden; some ambiguity around racemate-to-enantiomer transitions.
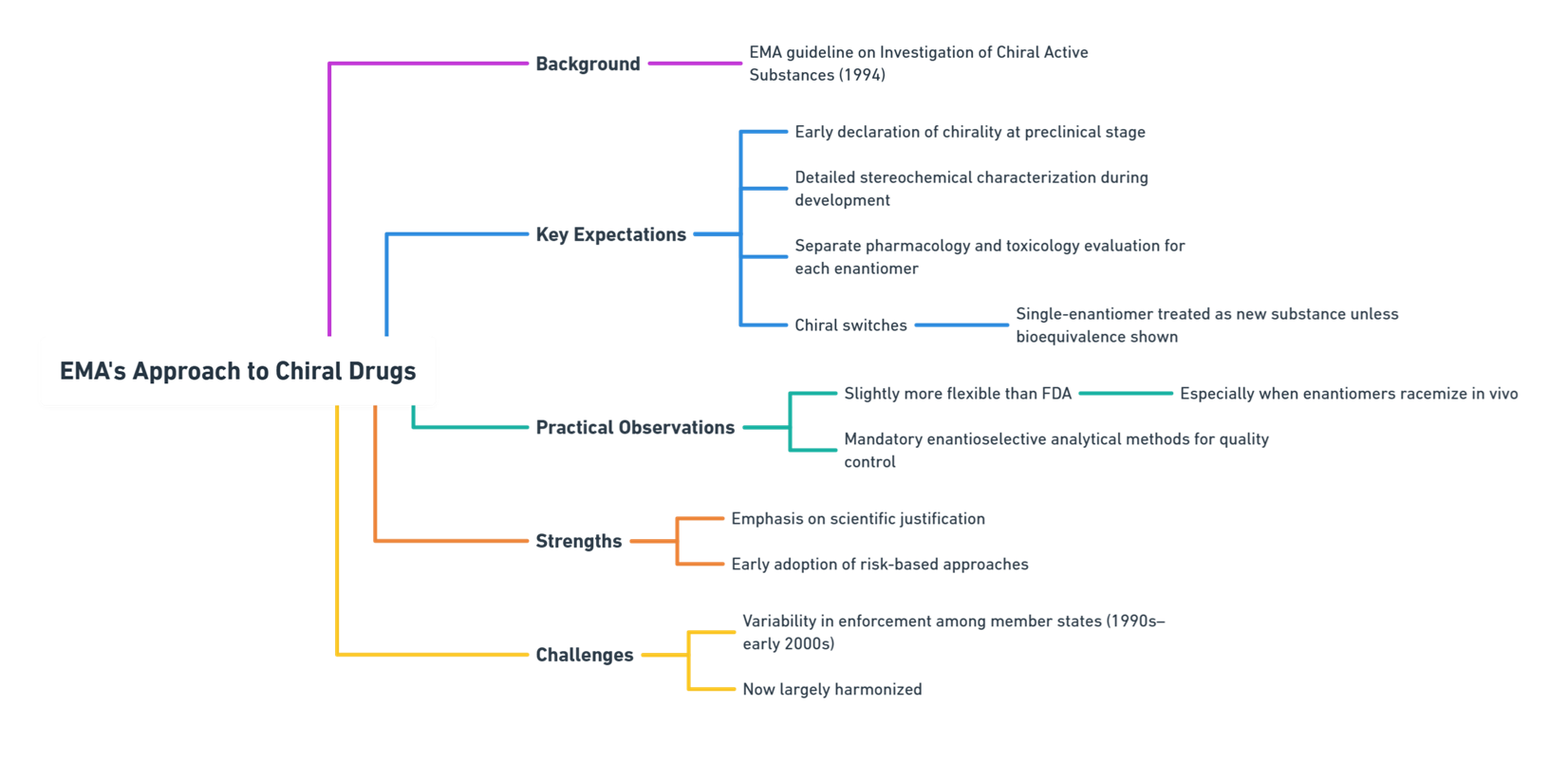
Europe: EMA’s Balance of Scientific Rigor and Pragmatism
The European Medicines Agency (EMA) followed with its 1994 Investigation of Chiral Active Substances guideline (EMA, 1994).
Key EMA Expectations
- Early declaration of chirality at the preclinical stage.
- Detailed stereochemical characterization during drug development.
- Separate evaluation of each enantiomer’s pharmacology and toxicology.
- Regulatory consideration for chiral switches: A single-enantiomer version must be treated as a new active substance unless bioequivalence is fully demonstrated.
Practical Observations
- EMA tends to be slightly more flexible than FDA regarding development paths, especially if enantiomers racemize in vivo.
- Enantioselective analytical methods are mandatory for quality control.
Strengths: Emphasis on scientific justification over rigid mandates; early adoption of risk-based approaches.
Challenges: Greater variability among member states in enforcement during the 1990s–early 2000s (now largely harmonized).
Japan: PMDA’s Cautious but Rigorous Evolution
Japan’s Pharmaceuticals and Medical Devices Agency (PMDA) aligned gradually with FDA/EMA standards through participation in ICH.
Key PMDA Expectations
- Mandatory characterization of chiral drugs at the earliest stage possible (Present State of New Chiral Drug Development, 2008).
- Separate pharmacokinetic and pharmacodynamic profiling for each enantiomer if technically feasible.
- Chiral-specific stability studies to assess in vivo or storage-related racemization.
Practical Observations
- Japan places particular emphasis on population-specific enantiomeric metabolism, reflecting ethnic factors in drug response (ICH E5, 1998).
- PMDA reviewers expect explicit discussion of how stereochemistry affects efficacy and safety in Japanese populations.
Strengths: Strong technical rigor; high-quality dossier expectations.
Challenges: Can be conservative in approving novel chiral switches without extensive local data.

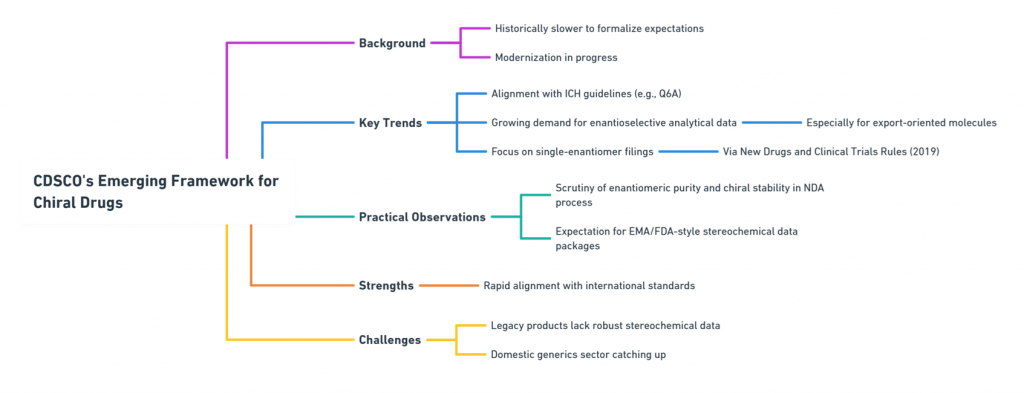
India: CDSCO’s Emerging Framework
India’s Central Drugs Standard Control Organization (CDSCO) has historically been slower to formalize specific expectations around chirality, but modernization is underway.
Key CDSCO Trends
- Alignment with ICH guidelines for new drug approvals, including Q6A specifications.
- Growing expectation for enantioselective analytical data, particularly for molecules intended for export markets.
- Increased focus on single-enantiomer drug filings through the New Drugs and Clinical Trials Rules, 2019 (CDSCO, 2019).
Practical Observations
- Enantiomeric purity and chiral stability are increasingly scrutinized during the New Drug Application (NDA) process.
- Indian regulators often expect developers to submit EMA- or FDA-style stereochemical data packages for new molecules.
Strengths: Rapid evolution toward international standards.
Challenges: Legacy products may not have robust stereochemical data; domestic generic sector still catching up.
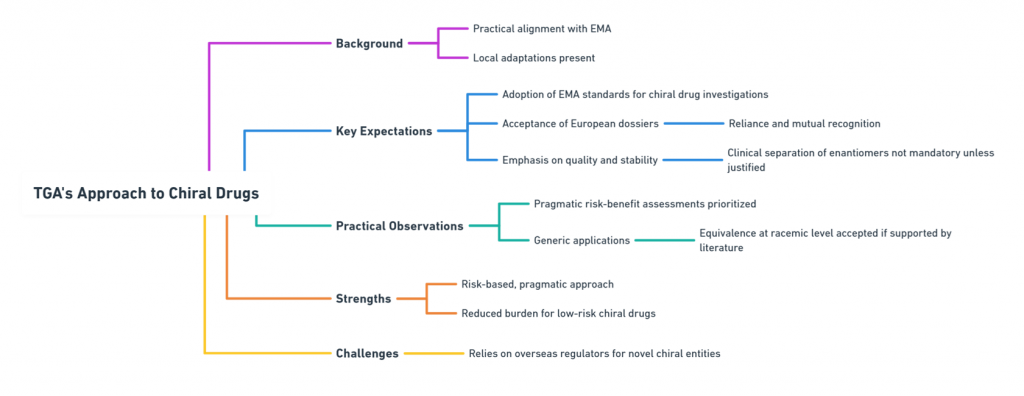
Australia: TGA’s Practical Alignment with EMA
Australia’s Therapeutic Goods Administration (TGA) largely mirrors EMA expectations, but with some unique local adaptations.
Key TGA Expectations
- Adoption of EMA’s chiral drug investigation standards.
- Acceptance of European dossiers via reliance and mutual recognition.
- Focus on quality and stability rather than mandatory clinical separation of enantiomers unless justified.
Practical Observations
- TGA emphasizes pragmatic risk-benefit assessment over rigid stereochemical purity targets.
- For generic applications, demonstration of equivalence at the racemic level may suffice if supported by literature.
Strengths: Pragmatic, risk-based approach; reduced burden for low-risk chiral drugs.
Challenges: Some reliance on overseas regulators for highly novel chiral entities.
Comparative Summary Table
| Agency | Key Chirality Focus | Chiral Switch Approach | Unique Feature |
|---|---|---|---|
| FDA | Full enantiomeric characterization and control | Treated as new drug; superiority proof expected | Strong stance on distinct evaluation |
| EMA | Scientific justification for choice of racemate/single | Treated as new entity unless bioequivalence proven | Flexibility in justification |
| PMDA | Stereochemistry tied to ethnic pharmacokinetics | Conservative; expects local data | Population-specific metabolism studies |
| CDSCO | Rapidly aligning with ICH standards | Increasing scrutiny; new rules emerging | Fast modernization |
| TGA | Follows EMA model pragmatically | Risk-based; some reliance on literature | Emphasis on practical risk assessment |
The Global Picture: More Aligned, Yet Still Distinct
Today, global regulatory expectations for chiral drugs are far more aligned than they were in the 1980s. ICH guidelines have created a common scientific backbone, and agencies often accept each other’s data through reliance pathways.
Yet, subtle differences persist:
- The degree of enforcement varies: FDA and PMDA tend to demand more standalone enantiomeric data.
- Ethnic considerations are emphasized more in Japan.
- Pragmatism vs. rigidity varies: TGA and, increasingly, CDSCO are more pragmatic; FDA is more rigid.
For multinational pharmaceutical companies, understanding these differences remains critical in designing global development strategies for chiral drugs.
Conclusion
While the importance of chirality is universally acknowledged, regulatory treatment of chiral drugs still reflects each agency’s culture, priorities, and risk tolerance. Recognizing these nuances is vital for developing, registering, and managing the lifecycle of chiral pharmaceuticals worldwide.
As we move forward, the next frontier lies not only in harmonizing standards but in anticipating how emerging scientific innovations will further challenge regulatory frameworks.
In the next episode, we will dive into how scientific and technological advances -from chiral separation to bioanalysis -have catalyzed the regulatory transformation.
What is in the next episode?
“Science fuels regulation -but how exactly? Join us in Episode 4: Technology as Catalyst: How Science Enabled the Regulatory Transformation.“
References
Agranat, I., Caner, H., & Caldwell, J. (2002). Putting chirality to work: The strategy of chiral switches. Nature Reviews Drug Discovery, 1(10), 753–768.
Central Drugs Standard Control Organization (CDSCO). (2019). New Drugs and Clinical Trials Rules. Government of India.
European Medicines Agency (EMA). (1994). Investigation of Chiral Active Substances (CPMP Note for Guidance 3CC29a).
Food and Drug Administration (FDA). (1992). Development of New Stereoisomeric Drugs (Policy Statement). Federal Register, 57(88), 22249-22250.
International Council for Harmonisation (ICH). (1998). E5(R1): Ethnic Factors in the Acceptability of Foreign Clinical Data.
International Council for Harmonisation (ICH). (1999). Q6A: Specifications – Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances.
Present State of New Chiral Drug Development and Review in Japan. (2008). Journal of Health Science, 54(1), 23–29.
Waldeck, B. (2003). Three-dimensional pharmacology: Effects of chirality on pharmacokinetics and pharmacodynamics. Pharmacology & Therapeutics, 95(1), 1–12.
Further Reading
