“When regulators caught up with science, a quiet molecular revolution began – changing the future of drug development forever.”
Introduction
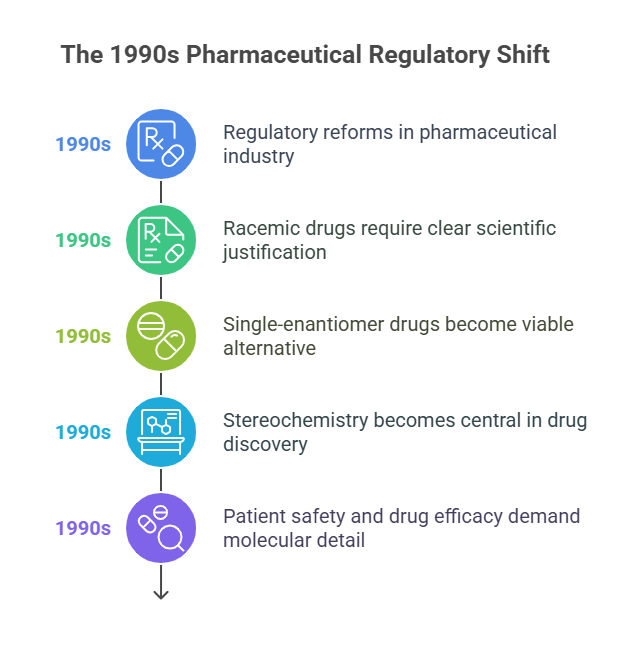
The 1990s represent a pivotal decade in pharmaceutical regulation, where long-overlooked nuances of molecular chirality finally received the attention they deserved. Before this era, enantiomers in drug substances were treated as interchangeable entities, with few regulatory frameworks requiring detailed evaluation of each stereoisomer. However, accumulating scientific evidence, technological advances, and a few high-profile pharmaceutical tragedies culminated in a historic regulatory reawakening.
This episode explores the critical regulatory milestones that redefined the standards for chiral drugs, focusing on landmark guidance from the United States Food and Drug Administration (FDA), the European Medicines Agency (EMA), and the burgeoning efforts toward international harmonization through the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). Together, these transformations laid the groundwork for how chiral drugs would be assessed, approved, and marketed into the twenty-first century.
Setting the Stage: Why the 1990s Were a Tipping Point
By the late 1980s, regulators could no longer ignore the growing body of evidence that enantiomers could have profoundly different biological properties. Academic journals increasingly reported enantioselective pharmacology, toxicology, and metabolism (Ariëns, 1984; 1986; 1987a,b,c; 1989.) Techniques like chiral chromatography and asymmetric synthesis had matured to the point where pharmaceutical companies could feasibly develop and characterize single-enantiomer drugs (Agranat, Caner, & Caldwell, 2002).
Concurrently, the globalization of pharmaceutical markets called for greater consistency in regulatory expectations. Different countries had varying requirements regarding drug characterization, and the lack of harmonized standards created confusion and inefficiency in drug development pipelines.
Moreover, ethical pressures mounted. Public health disasters such as the thalidomide tragedy in the 1960s, though decades past, remained a sobering reminder of the catastrophic consequences of overlooking molecular details (McBride, 1961). The scientific community increasingly argued that regulators bore a responsibility to demand more rigorous stereochemical evaluation. Thus, the 1990s became the decade when regulatory authorities around the world decided to act decisively.
The U.S. FDA’s Landmark 1992 Policy Statement
In May 1992, the U.S. FDA issued its seminal Policy Statement for the Development of New Stereoisomeric Drugs (FDA, 1992). Published in the Federal Register and simultaneously shared in the academic journal Chirality, the document signaled a seismic shift in regulatory expectations.
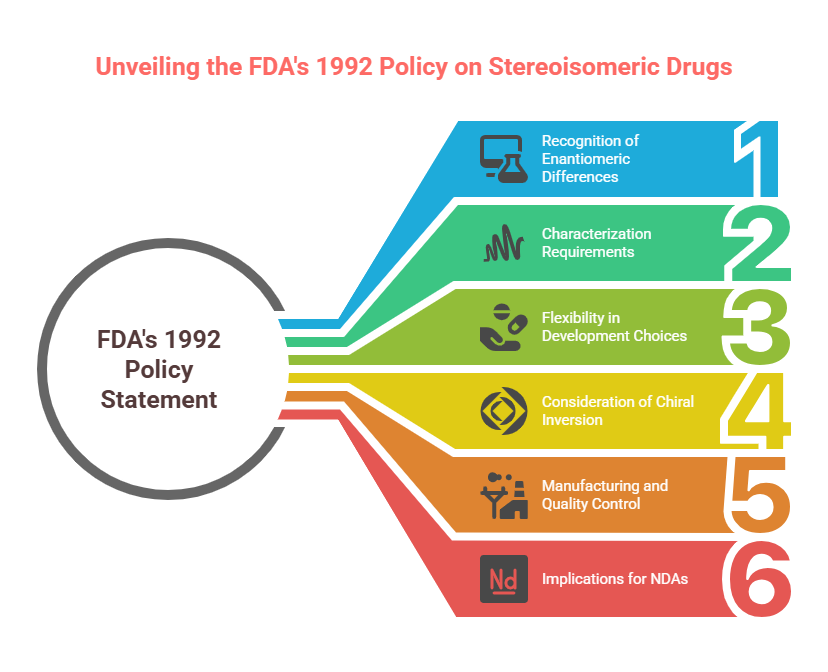
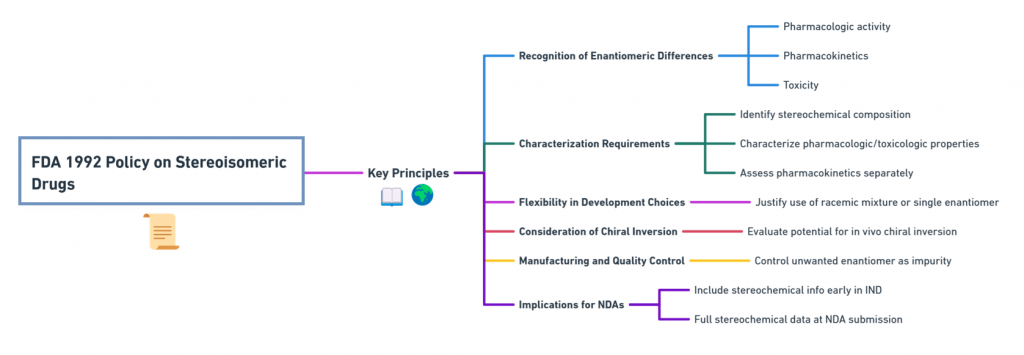
Key Principles of the FDA 1992 Policy
- Recognition of Enantiomeric Differences
The FDA explicitly acknowledged that enantiomers could differ significantly in pharmacologic activity, pharmacokinetics, and toxicity. Therefore, each enantiomer must be considered as a distinct chemical entity for regulatory purposes unless proven otherwise. - Characterization Requirements
Drug developers were expected to:- Identify the stereochemical composition of drug substances.
- Characterize the pharmacologic and toxicologic properties of each enantiomer.
- Assess pharmacokinetics separately for each enantiomer when possible.
- Flexibility in Development Choices
The policy did not mandate the development of single-enantiomer drugs. Rather, it required a justification for choosing either a racemic mixture or a single enantiomer, based on scientific evidence. - Consideration of Chiral Inversion
Developers needed to evaluate the potential for in vivo chiral inversion—conversion of one enantiomer into its mirror image within the body. - Manufacturing and Quality Control
For single-enantiomer drugs, manufacturers were required to specify and control the presence of the unwanted enantiomer, treating it akin to an impurity. - Implications for New Drug Applications (NDAs)
Sponsors were advised to include stereochemical information early in Investigational New Drug (IND) submissions and ensure full stereochemical data was available at the time of NDA submission.
Why It Was Revolutionary
The 1992 policy represented the first formal acknowledgment by a major regulatory agency that chirality was not a minor technicality but a fundamental aspect of drug efficacy and safety. It also codified the expectation that stereochemistry should be addressed early—not retrofitted at late stages of development. By issuing this policy, the FDA effectively ended the era when developers could overlook enantiomeric properties without consequence
The EMA’s 1994 “Investigation of Chiral Active Substances” Guideline
Following the FDA’s lead, the European Medicines Agency (EMA) -then still emerging from its predecessor agencies -released its Note for Guidance: Investigation of Chiral Active Substances in 1994 (EMA, 1994).
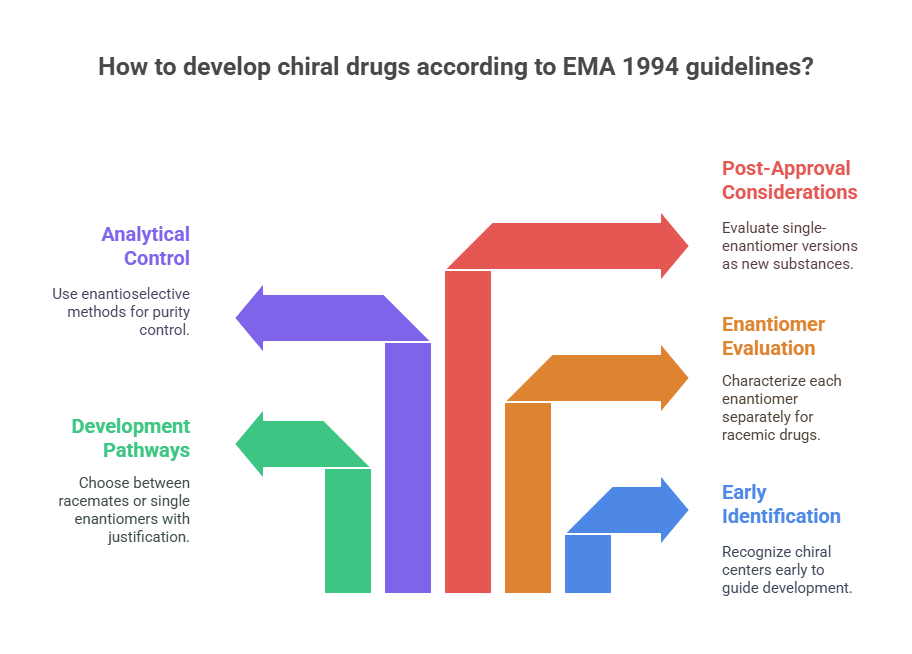
Highlights of the EMA 1994 Guideline
- Early Identification of Chirality
Applicants were expected to recognize and declare chiral centers at the earliest stages of development. - Development Pathways for Racemates vs. Single Enantiomers
The EMA permitted the development of either racemates or single enantiomers, but required scientific justification for the chosen path. - Separate Evaluation of Enantiomers
For racemic drugs, the pharmacodynamic, pharmacokinetic, and toxicological profiles of each enantiomer needed to be characterized individually wherever possible. - Analytical Control
The guideline emphasized the need for enantioselective analytical methods to control enantiomeric purity throughout development and manufacturing. - Post-Approval Considerations
The EMA raised awareness that if a company intended to develop a single-enantiomer version of an already approved racemate (a “chiral switch”), it would need to be evaluated as a new active substance.
Harmonizing with FDA – But Subtly Different
While broadly aligned with the FDA’s policy, the EMA’s guidance reflected subtle European nuances:
- A stronger emphasis on detailed justification for developing racemates.
- Slightly more caution around approving chiral switches without demonstrated clinical advantage.
- A consistent push towards quality-by-design concepts, even in stereochemical decisions.
Japan’s Parallel Movement: PMDA’s Informal Alignment
Japan’s Pharmaceuticals and Medical Devices Agency (PMDA) did not issue a standalone chirality guidance in the early 1990s. However, through participation in ICH discussions, Japanese regulators informally aligned with FDA and EMA expectations (Present State of New Chiral Drug Development and Review in Japan, 2008). New drug applications (NDAs) in Japan began including detailed enantiomeric characterization data. In practice, Japan required:
- Early identification of stereochemistry.
- Enantiomer-specific pharmacokinetic and pharmacodynamic studies where feasible.
- Enantiomeric purity control per ICH standards.
Thus, while Japan’s policy evolution was quieter, it was no less impactful.
The Role of ICH: Globalizing Stereochemical Standards
The 1990s also witnessed the birth of the ICH -a tripartite initiative among the United States, Europe, and Japan aimed at harmonizing drug development standards.
Two key ICH guidelines integrated stereochemistry into global expectations:
- ICH Q6A (1999): This guideline on specifications explicitly addressed how to handle enantiomeric impurities, requiring that the presence of undesired enantiomers be controlled using validated chiral analytical methods (ICH, 1999).
- ICH E5 (1998): Though primarily about ethnic factors in clinical data, E5 emphasized the importance of understanding metabolism differences -including enantiomeric differences -across populations.
Through ICH, the stereochemical policies first introduced by the FDA and EMA became international norms, extending their influence far beyond the U.S. and Europe.
Industry Response: A Paradigm Shift
The pharmaceutical industry rapidly adapted to the new regulatory climate:
- Development pipelines changed: More drugs were designed as single-enantiomer products from the outset.
- Manufacturing evolved: Investment in asymmetric synthesis, chiral resolution techniques, and chiral analytics soared.
- Patent strategies shifted: Companies began exploring “chiral switches” as a way to extend product lifecycles (Agranat & Wainschtein, 2010).
This transformation was not merely reactive; many firms recognized that a purer, more targeted drug profile could offer genuine therapeutic benefits and marketing advantages.
The Legacy of the 1990s Shift
The regulatory reforms of the 1990s permanently changed the pharmaceutical landscape:
- Racemic drugs could still be developed, but only with clear scientific justification.
- Single-enantiomer drugs became a viable -and often preferable -alternative.
- Stereochemistry moved from an afterthought to a central consideration in drug discovery and development.
Perhaps most importantly, regulators signaled that patient safety and drug efficacy demanded attention to molecular detail. Chirality was no longer a boutique scientific interest; it was a regulatory imperative.
Conclusion
The 1990s marked the end of regulatory ambivalence toward chirality and the dawn of a more nuanced, scientifically rigorous approach to stereochemistry in drug development. The FDA’s 1992 policy statement, the EMA’s 1994 guidance, and the ICH’s global initiatives collectively ushered in a new era -one where mirror images could no longer be treated as equivalent.
In the next episode, we will explore how different regulatory agencies worldwide adopted and adapted these new standards, and where subtle differences in approach still exist.
What is in the next episode?
“How do FDA, EMA, PMDA, CDSCO, and TGA really compare on chiral drug regulation? Find out in Episode 3: Beyond Borders: Comparing How Global Agencies Regulate Chiral Drugs.“
References
Ariens, E. J. (1984). “Stereochemistry, a basis for sophisticated nonsense in pharmacokinetics and clinical pharmacology”. European Journal of Clinical Pharmacology. 26 (6): 663–668. doi:10.1007/bf00541922.
Ariëns, E.J. (1986). “Chirality in bioactive agents and its pitfalls”. Trends in Pharmacological Sciences. 7: 200–205. doi:10.1016/0165-6147(86)90313-5.
- Ariëns, Everardus J. (1987a). “Stereochemistry in the analysis of drug-action. Part II”. Medicinal Research Reviews. 7 (3): 367–387. doi:10.1002/med.2610070305.
Ariëns, E.J (1987b). “Implications of the Neglect of Stereochemistry in Pharmacokinetics and Clinical Pharmacology”. Br. J. Clin. Pharmacol. Ther. 21 (10): 827–829. doi:10.1177/106002808702101013.
- Ariëns, Everd J. (1987c). “Implications of the Neglect of Stereochemistry in Pharmacokinetics and Clinical Pharmacology”. Drug Intelligence & Clinical Pharmacy. 21 (10): 827–829. doi:10.1177/106002808702101013.
Ariëns, E. J. (1989). “Stereoselectivity in the Action of Drugs”. Pharmacology & Toxicology. 64 (4): 319–320. doi:10.1111/j.1600-0773.1989.tb00655.x. .
Agranat, I., Caner, H., & Caldwell, J. (2002). Putting chirality to work: The strategy of chiral switches. Nature Reviews Drug Discovery, 1(10), 753–768.
Agranat, I., & Wainschtein, S. R. (2010). The strategy of enantiomer patents of drugs. Drug Discovery Today, 15(5-6), 163–170.
European Medicines Agency (EMA). (1994). Investigation of Chiral Active Substances (CPMP Note for Guidance 3CC29a).
Food and Drug Administration (FDA). (1992). Development of New Stereoisomeric Drugs (Policy Statement). Federal Register, 57(88), 22249-22250.
International Conference on Harmonisation (ICH). (1999). Q6A: Specifications – Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances.
McBride, W. G. (1961). Thalidomide and congenital abnormalities. The Lancet, 278(7216), 1358.
Present State of New Chiral Drug Development and Review in Japan. (2008). Journal of Health Science, 54(1), 23–29.
Waldeck, B. (2003). Three-dimensional pharmacology: Effects of chirality on pharmacokinetics and pharmacodynamics. Pharmacology & Therapeutics, 95(1), 1–12.
Further Reading
